41
VROZENÉ PORUCHY KRVETVORBY A JEJICH MANIFESTACE V DOSPĚLÉM VĚKU Jaroslav Čermák Ústav hematologie a krevní transfuze, Praha
| Date post: | 07-Apr-2019 |
| Category: |
Documents |
| Upload: | hoangkhanh |
| View: | 215 times |
| Download: | 0 times |

VROZENÉ PORUCHY
KRVETVORBY A JEJICH MANIFESTACE V DOSPĚLÉM VĚKU
Jaroslav Čermák
Ústav hematologie a krevní transfuze, Praha

VROZENÉ SYNDROMY SELHÁNÍ KRVETVORBY

VROZENÉ SYNDROMY SELHÁNÍ KRVETVORBY

VROZENÉ SYNDROMY SELHÁNÍ KRVETVORBY

VROZENÉ SYNDROMY SELHÁNÍ KRVETVORBY

VROZENÉ SYNDROMY SELHÁNÍ KRVETVORBY

PORUCHY REPARACE DNA
Fanconiho anémie mutace genů FANC komplexu (zásadní role v reparaci poškozené DNA) incidence 0.1-0.5/ 100.000 narozených dědičnost autosomálně recesivní či vázaná na X chromozóm výrazně heterogenní onemocnění manifestace selhání kostní dřeně – většinou kolem 7.roku věku : trombocytopenie a makrocytóza progredující pancytopenie, vysoké riziko vzniku malignit (50x vyšší než u zdravé populace) vrozené anomálie u 60% nemocných (skelet, malý vzrůst, malformace GIT, urogenitálního traktu, neurologické abnormity)

PORUCHY REPARACE DNA

PORUCHY REPARACE DNA

PORUCHY REPARACE DNA
Fanconiho anemie ♀ 23 let, výška 128 cm
Anemie s výraznou makrocytozou a
normálním počtem retikulocytů
Leukopenie s granulocytopenií
Výrazná trombocytopenie

FANCONIHO ANEMIE → MDS
Cytogenetika : 46,XX
(při kontrole susp. monosomie 7)
Molekulární
genetika :
není mutace TP53 není mutace BRCA2
prokázána mutace FANCA genu (homozygot c.3403_c.3105delTTC)

FANCONIHO ANEMIE → MDS
Fanconiho anemie ♀ 23 let, výška 128 cm
Dg. : Myelodysplastický syndrom typu RCMD – vývojem z Fanconiho anemie (potvrzené molekulárně genetickým vyšetřením)
Klinický průběh : anemie s transfuzní dependencí (1-2 TU za 6 týdnů) občasné krvácivé projevy → postupný pokles počtu trombocytů (25-30 tis.)
Léčba: alogenní SCT od HLA identické sestry (bez přítomnosti mutace FANCA genu) nyní D 120 po SCT – přihojení štěpu, obnova dárcovské krvetvorby

PORUCHY TVORBY RIBOSOMŮ

PORUCHY TVORBY RIBOSOMŮ

PORUCHY TVORBY RIBOSOMŮ

PORUCHY TVORBY RIBOSOMŮ

PORUCHY TVORBY RIBOSOMŮ
Shwachman Diamondův syndrom
• pancytopenie se známkami selhání kostní dřeně a rozvojem myelodysplastického syndromu
Mutace SBDS genu : nutný pro normální metabolismus RNA, funkci a vazbu ribosomálních podjednotek.

PORUCHY TVORBY RIBOSOMŮ Shwachman Diamondův syndrom
NEMOCNÝ ♂ 39 let
retardace růstu
atrofie pankreatu s lipomatozou selhání kostní dřeně
ÚHKT : prokázána mutace SBDS genu
Dg.: sledován na Dětské hematologické klinice a OKH FN Motol s dg. susp. Shwachman Diamondův syndrom, neléčen, postupně progredující leukopenie a trombocytopenie

PORUCHY TVORBY RIBOSOMŮ
Shwachman Diamondův syndrom
Pancytopenie s přítomností blastů
v periferní krvi
♂ 39 let, Dg:
MDS RCMD
Kostní dřeň normocelulární,
dysplasie ve všech řadách 13 % myeloblastů

PORUCHY TVORBY RIBOSOMŮ
Shwachman Diamondův syndrom CYTOGENETIKA

PORUCHY TVORBY RIBOSOMŮ
Shwachman Diamondův syndrom ♂ 39 let, výška 146 cm
Dg. : Myelodysplastický syndrom typu RAEB-2 až RAEB-T– vývojem z Shwachman Diamondova syndromu (potvrzeno molekulárně genetickým vyšetřením)
Klinický průběh : progredující cytopenie s nutností antibiotické profylaxe, dependence na transfuzích trombocytů, postupný nárůst počtu blastů v kostní dřeni
Léčba: kombinovaná chemoterapie, dosažena CR, alogenní SCT od HLA identického nepříbuzného dárce, přihojení štěpu, po 3.měsících obnova smíšené chiméry → redukce imunosuprese, přechodná stabilizace nálezu, následný relaps → AML rezistentní na léčbu, + D 270 po SCT

PORUCHY REGULAČNÍCH GENŮ
GATA-2 : „zinc-finger“ transkripční faktor – gen lokalizován na chromozomu 3
Funkce :
regulátor efektivní genové exprese v hemopoetických kmenových
buňkách – ovlivnění úrovně proliferace a diferenciace HSC.
Exprese GATA- regulována epigenetickou modifikací.
Genetický defekt GATA-2 :
imunodeficience a predispozice k MDS a AML /MonoMAC syndrom/.
RCC – refrakterní dětská cytopenie :
u některých nemocných nalezena defektní exprese GATA-2 → může predisponovat
k následným ( „second hit“) mutacím vedoucím ke klonální proliferaci.

PORUCHY REGULAČNÍCH GENŮ
NEMOCNÝ ♂ 19 let
Dg. - MonoMAC syndrom :
autosomálně dominantní syndrom – mnohočetné mutace GATA2 genu monocytopenie deficit NK a B lymfocytů recidivující infekty – mykosy, mykobakteria plicní alveolární proteinoza
Nemocný : Recidivující plicní mykotické infekce + incipientní alveolární proteinoza
Absolutní monocytopenie

PORUCHY REGULAČNÍCH GENŮ
NEMOCNÝ ♂ 19 let
Průběh onemocnění :
během 6 měsíců – progredující cytopenie : recidivující infekty - plíce trvalá antibiotická profylaxe závislost na transfuzích RBC a PLT
KO : pancytopenie s těžkou neutropenií

PORUCHY REGULAČNÍCH GENŮ

PORUCHY REGULAČNÍCH GENŮ
Kostní dřeň : normocelulární, dysplasie ve všech řadách 3 % myeloblastů
Cytogenetika : 46 ,XY 45,XY,-7

PORUCHY REGULAČNÍCH GENŮ
MonoMAC syndrom ♂ 19 let
Dg. : Myelodysplastický syndrom typu RCMD– vývojem z MonoMAC syndromu (potvrzeno molekulárně genetickým vyšetřením), monosomie 7.chromosomu
Klinický průběh : progredující pancytopenie s nutností antibiotické profylaxe, dependence na transfuzích trombocytů a erytrocytů, recidivující infekty - plíce
Léčba: alogenní SCT od HLA identického nepříbuzného dárce nyní D 420 po SCT – přihojení štěpu, bez komplikací.

VROZENÉ SYNDROMY SELHÁNÍ KRVETVORBY

PORUCHY TVORBY HEMOGLOBINU
Nemocný ♂ 28 let, hypochromní mikrocytární anemie
Dg. : mikrocytární anémie od dětství Hb v rozmezí 90-120 g/l, spontánní výkyvy.
Anamn: nemocný je Řek, u obou rodičů údajně zjištěna thalasemie. KO : hypochromní mikrocytární anémie normální počet RBC, RTC, ↑ RDW
? thalasemie

PORUCHY TVORBY HEMOGLOBINU Otec nemocného : ♂ 57 let
KO : bez zjevné patologie
Analýza globinových genů: heterozygotní mutace 1 páru α genů ( - α3,7 ) - mutace polyA signálu – snížená stabilita mRNA heterozygotní forma α+ thalasemie
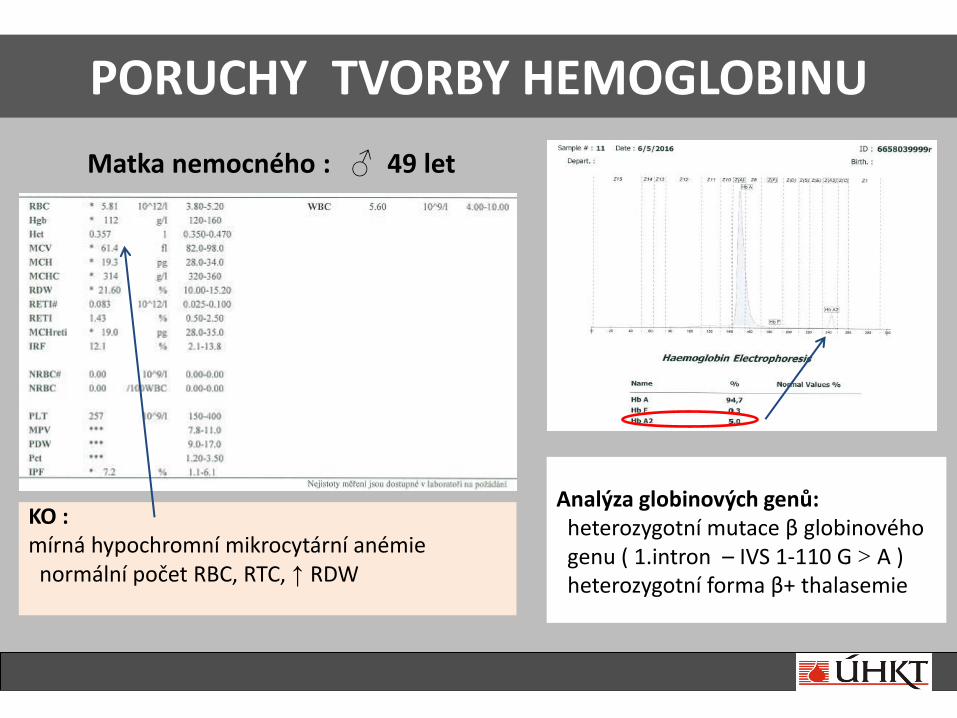
PORUCHY TVORBY HEMOGLOBINU
Matka nemocného : ♂ 49 let
KO : mírná hypochromní mikrocytární anémie normální počet RBC, RTC, ↑ RDW
Analýza globinových genů: heterozygotní mutace β globinového genu ( 1.intron – IVS 1-110 G > A ) heterozygotní forma β+ thalasemie

PORUCHY TVORBY HEMOGLOBINU Nemocný ♂ 28 let
Analýza globinových genů: neprokázána mutace ani α globinového, ani β globinového genu Nemocný nemá thalasemii (?!)

PORUCHY TVORBY HEMOGLOBINU Nemocný ♂ 28 let
Analýza globinových genů: neprokázána mutace ani α globinového, ani β globinového genu Nemocný nemá thalasemii (?!) Nemocný má : - hypochromii a mikrocytózu, normální počet RBC - spontánní výkyvy hodnot Hb - extremní anizocytózu (↑↑ RDW ) - známky přetížení Fe : sFe - 26.1 mmol/l, sferitin – 867 μg/l ( dostal 1 TU erymasy před 15 lety ) Jak postupovat dále v dg. ?

PORUCHY TVORBY HEMOGLOBINU Nemocný ♂ 28 let - kongenitální sideroblastická anémie
Buněčně bohatá dřeň s hyperplasií červené řady, mírnou dysplasií a četnými sideroblasty (sideroblasty: 93%, 16% prsténčité formy).
R452H
Delta amino levulát syntetáza :
Mutace exonu 9 G→A Arg452His

PORUCHY TVORBY HEMOGLOBINU

PORUCHY TVORBY HEMOGLOBINU
OTEC :
mikrocytóza, hypochromie, bez anémie
Heterozygotní mutace : CD142 TAA>▐AA[Stop>Gln]
Hb Constant Spring CS α/ α α

PORUCHY TVORBY HEMOGLOBINU
MATKA :
mikrocytóza, hypochromie, polyglobulie
Heterozygotní mutace --SEA zahrnující geny HBA1 i HBA2
α α /--SEA

PORUCHY TVORBY HEMOGLOBINU
NEMOCNÁ :
HbH disease mutace 3 alfa globinových genů
CSα/--SEA Klinicky : thalasemia intermedia až major s transfuzní dependencí Léčba: chelatace, SCT (?)

VROZENÉ PORUCHY KRVETVORBY
Vrozené poruchy krvetvorby
výrazný nárůst v posledních 10 letech : a) zlepšená diagnostika u dětí (molekuárně genetické metody) b) zlepšená diagnostika subklinických forem, které se mohou projevit až v období dospívání c) narůstající migrace populace (alfa thalasemie, srpkovitá anemie) Diagnostika a povědomí nemocných o jejich onemocnění jsou důležité pro prevenci vzniku homozygotních forem či kombinovaných heterozygotních poruch s těžkým klinickým průběhem.

VROZENÉ PORUCHY KRVETVORBY Poděkování
Centrum vzácných chorob krvetvorby Praha ÚHKT Mgr. Monika Beličková MVDr. Svatopluk Škranc MUDr. Dana Mikulenková MUDr. Radka Šimečková RNDr. Jana Březinová, PhD Dětská hematologická a dětská onkologická klinika FN Praha Motol Prof. MUDr. Jan Starý, DrSc. & Prof. MUDr. Dagmar Pospíšilová, CSc. Dětská klinika FN Olomouc a Centrum vzácných chorob krvetvorby Olomouc









