Protokoly a návody pro praktikum Metody molekulární biologie v rostlinné ekologii a systematice Laboratoř molekulární biologie rostlin, PřF JCU, Branišovská 31, České Budějovice 2008, Miroslava Herbstová, Petr Koutecký & Jiří Košnar
Transcript
Protokoly a návody pro praktikum
Metody molekulární biologie v rostlinné ekologii
a systematice
Laboratoř molekulární biologie rostlin, PřF JCU, Branišovská 31, České Budějovice
2008, Miroslava Herbstová, Petr Koutecký & Jiří Košnar
Základní zásady práce v molekulárně biologické laboratoři
• při práci s DNA pracujeme pouze se sterilními autoklávovanými špičkami a zkumavkami
(v krabičkách a kádinkách označených páskou s černými pruhy), kontaminovaný materiál odhazujeme do určených nádob
• pro PCR, ředění DNA a primerů používáme výhradně sterilní vodu (autoklávovaná Milli-Q voda nebo koupená voda od f. Top-Bio)
• pro omývání laboratorního nádobí používáme destilovanou vodu se savem
• s roztokem Syber Greenu (nanášecí pufr, LB = loading buffer), který je potencionální mutagen, a s veškerými věcmi, které s ním přicházejí do styku (elektroforetické vany, hřebínky, pipeta na nanášení vzorků…), pracujeme v rukavicích, kontaminovanými rukavicemi se nedotýkáme ničeho jiného, gely vyhazujeme zabalené v papíru do odpadkového koše k tomu určeného
• akrylamid je neurotoxin, dbáme zvýšené opatrnosti, pracujeme zásadně v rukavicích, kontaminovaný materiál a gely vyhazujeme do zvláštní nádoby, zásobní roztoky uchováváme v ledničce (2-8 °C)
• dbáme zvýšené opatrnosti při práci s elektrickým proudem (elektroforéza apod.)
• s těkavými chemikáliemi (chloroform, isoamylalkohol, isopropanol, kyselina octová, TEMED, kyselina chlorovodíková…) pracujeme zásadně v digestoři se zapnutým odtahem
• s DNA a většinou dalších biochemikálií (enzymy, primery…) pracujeme výhradně na ledu, zvlášť citlivé jsou enzymy (polymeráza, restrikční enzymy ad.) a některé pufry
• pokud odcházíme z laboratoře poslední (i jen na chvíli), vždy zamkneme
Obsah
Metody analýzy DNA
Metody izolace DNA.............................................................................................................1
A) Izolace DNA z čerstvého nebo sušeného rostlinného materiálu s použitím Invisorb Spin Plant Mini Kit (INVITEK) ............................................................................................1
B) Extrakce DNA ze sušeného rostlinného materiálu – CTAB metoda...............................3 C) Extrakce DNA pomocí NaOH.......................................................................................4 D) Měření koncentrace DNA a ředění DNA.......................................................................5 E) Otestování kvality DNA na agarosovém gelu ................................................................5
Polymerázová řetězová reakce ........................................................................................... 6
PCR-RFLP (Restriction Fragment Lenght Polymorphism).............................................16
AFLP (Amplified Fragment Length Polymorphism)........................................................17
A) Protokol s využitím AFLP kitů od firmy Invitrogen ....................................................18 B) Protokol bez využití AFLP kitů ...................................................................................20 C) Přesrážení octanem sodným ........................................................................................23
Horizontální agarosová elektroforéza ...............................................................................24 Vertikální polyakrylamidová elektroforéza na 6,2% kontinuálním gelu............................25 Práce s dokumentačním systémem Gel Imager a softwarem Scion VisiCapture................26
Isozymy
Princip metody.................................................................................................................27 Histochemické barvení.....................................................................................................28 Základy interpretace isozymových patterns .....................................................................29 Extrakce vzorků ...............................................................................................................30 Elektroforéza a barvení ....................................................................................................31 Vysušení gelu ..................................................................................................................36 Jednotlivé enzymové systémy ..........................................................................................37
Obsluha přístrojů
Měření koncentrace DNA pomocí spektrofotometru Biowave II ......................................44 Zacházení s gradientovým termocyclerem TC-XP Bioerg ................................................45 Zacházení s termocyclerem Biometra T3000 se třemi bloky.............................................46 Měření na pH-metru Hanna pH 211 .................................................................................47
A) Izolace DNA z čerstvého nebo sušeného rostlinného materiálu s
použitím Invisorb Spin Plant Mini Kit (INVITEK)
Princip
Metoda využívá unikátní patentované technologie Invisorb®. Umožňuje rychlou a účinnou izolaci kvalitní genomické DNA z čerstvého, mraženého nebo suchého rostlinného materiálu (např. z listů, kořenů, plodů nebo semen), také z řas a sinic. Podstatou je interakce mezi negativně nabitými fosfátovými skupinami DNA a pozitivně nabitými skupinami na povrchu membrány v kolonce, kde se DNA váže a po přečištění se uvolní vhodným elučním pufrem. Vazba nebo naopak uvolnění DNA z kolonky závisí na koncentraci solí a pH použitých pufrů.
Postup
Obecné
• Před použitím promývacích pufrů (Wash Buffer I a II) zkontrolujeme přidání ethanolu (zaškrnutí příslušného políčka na láhvi).
• K času pro centrifugaci přičítáme čas nutný k dosažení požadovaných otáček.
• Odpadní filtráty vzniklé během izolace vyléváme do připravené kádinky.
• Proteinasa K je součástí kitu. Dodává se se v lyofilizovaném stavu. Před prvním použitím přidáme 1 ml sterilní vody a rozpustíme převracením zkumavky v ruce, krátce stočíme.
• Před vlastní prací odpipetujeme do 1,5 ml eppendorfky potřebné množství elučního pufru (Ellution buffer D) a předehřejeme v termobloku na 65°C.
Homogenizace
• V tekutém dusíku vymrazíme 1,5 ml eppendorfku s vloženým rostlinným materiálem (cca 60 mg rostlinné tkáně v čerstvém stavu nebo odpovídající množství sušeného materiálu). Vymrazíme homogenizátorek a krouživými pohyby drtíme vzorek ve vymražené eppendorfce.
• Jinou možností je drtit vzorek ve vymražené třecí misce. Rozdrcený materiál vymraženým skalpelem převedeme do připravené eppendorfky.
• K drcení vzorku je možné použít také mořský písek.
• V některých případech je možné drtit přímo čerstvý nebo suchý materiál za pokojové teploty přímo v lyzačním pufru (Lysis buffer P) bez použití kapalného dusíku.
Lýza
• K rozdrcenému materiálu přidáme 400 µl lyzačního pufru (Lysis buffer P) a 20 µl
proteinázy K (rozštěpení proteinů, inaktivace DNas). Lyzační pufr a proteinasu přidáváme dříve než vzorek zcela rozmrzne.
• Vortexujeme a necháme 30 min nebo déle inkubovat při 65°C. Během inkubace 2–3× promícháme.
• Připravíme si kolonky (Spin Filter) do 2,0 ml sběrných zkumavek (Receiver Tube).
Izolace DNA
2
Filtrace
• Lyzační směs přeneseme na kolonku (nejen roztok, ale i rozdrcenou tkáň). Centrifugujeme 1 min při 12 000 rpm (dle potřeby déle). Kolonky vyhodíme.
Odstranění RNA
• Pokud je potřeba pro další analýzy odstranit RNA, přidáme 5–40 µl RNAasy A (firma
Promega; 10mg/ml; není součástí kitu). Množství závisí na typu a výchozí navážce materiálu.
• Vortexujeme a necháme 5 min inkubovat při pokojové teplotě.
Navázání DNA
• Přidáme 200 µl Binding Buffer P a vortexujeme.
• Umístíme nové kolonky do 2,0 ml sběrných zkumavek. Suspenzi přeneseme na kolonku a inkubujeme 1 min.
• Centrifugujeme 1 min při 12 000 rpm.
Promytí I
• Odstraníme filtrát a kolonky umístíme zpět do 2,0 ml sběrných zkumavek.
• Přidáme 550 µl promývacího pufru I (Wash Buffer I) a centrifugujeme 1 min při 12 000 rpm.
• Odstraníme filtrát a kolonku umístíme zpět do 2,0 ml sběrné zkumavky.
Promytí II
• Přidáme 550 µl promývacího pufru II (Wash Buffer II) a centrifugujeme 1 min při
12 000 rpm.
• Odstraníme filtrát a kolonku umístíme zpět do 2,0 ml sběrné zkumavky.
• Tento krok opakujeme ještě jednou.
Vysušení
• Pro odstranění zbytků ethanolu z promývacího pufru centrifugujeme 2 min při 12 000 rpm.
Eluce
• Kolonky umístíme do sterilních 1,5 ml eppendorfek (nejsou součástí kitu). Pokud předpokládáme delší skladování DNA, použijeme tzv. safe-lock eppendorfky.
10–25 min při pokojové teplotě v uzavřené digestoři bez odtahu.
• Centrifugujeme 1 min při 10 000 rpm. Pozn.: Při centrifugaci nelze zavřít víčka. Při vkládání eppendorfek do centrifugy směrujeme otevřená víčka ve směru otáčení rotoru, abychom zabránili ulámání víček během centrifugace.
• Pro získání maximálního výtěžku DNA je možné eluovat nejprve 50 µl elučního pufru a
v druhém kroku dalšími 50 µl elučního pufru. Pokud nám nejde o celkový výtěžek DNA,
ale o co nejvyšší koncentraci DNA, pak k eluci v druhém kroku použijeme první eluát (tj. roztok DNA pipetujeme znovu na kolonku). Inkubujeme 5 - 10 min a centrifugujeme.
Izolace DNA
3
B) Extrakce DNA ze sušeného rostlinného materiálu – CTAB
metoda
Princip
Metoda je založena na schopnosti CTAB (cetyltrimetylamoniumbromid) vytvářet komplex s nukleovými kyselinami, který je při vysoké koncentraci solí rozpustný (0,7M NaCl), ale při snížené koncentraci solí (0,45M NaCl) vytváří sraženinu. CTAB zároveň působí jako detergent, který uvolňuje DNA z membrán a proteinů. Na základě rozdílné rozpustnosti CTAB v porovnání s DNA je lze oddělit a získat dostatečně čistou rostlinnou DNA.
Tuto metodu lze použít k izolaci většího množství DNA.
Postup
Homogenizace
• Viz extrakce DNA kitem Invisorb Spin Plant Mini Kit (INVITEK).
Vlastní extrakce DNA
• K rozdrcenému materiálu přidáme 700 µl roztoku CTAB a 10 µl 2-merkaptoethanolu. Pracujeme v digestoři.
• Zkumavky uzavřeme, krátce promícháme na vortexu a inkubujeme 30 min v termobloku při 60 °C.
• Během prvních minut inkubace přidáme ke každému vzorku „na špičku špachtle“ polyvinylpyrolidonu (PVP).
• Po inkubaci přidáme 500 µl směsi chloroform: isoamylalkohol (24:1). Pracujeme v digestoři
• Dobře uzavřené zkumavky 2–3× převrátíme a necháme cca 5 min stát.
• Centrifugujeme 10 min při 13 800 rpm.
• Supernatant (cca 500 µl) opatrně přepipetujeme do nových označených 1,5 ml eppendorfek. Je třeba dát pozor, abychom pipetovali jen průhledný supernatant. Pracujeme stále v digestoři a použité špičky a eppendorfky vyhazujeme do zvláštní nádoby na nebezpečný odpad.
• Přidáme 500 µl vychlazeného isopropanolu (z mrazáku).
• 1–2 × převrátíme (netřepeme) a necháme cca 30 min stát v –20 °C.
• Centrifugujeme 5 min při 13 800 rpm.
• Supernatant opatrně slijeme do kádinky (v digestoři), na dně eppendorfky lze vidět drobný matně bílý pelet DNA. Otevřené eppendorfky převrátíme dnem nahoru na filtrační papír (buničinu) pro odstranění zbytku kapaliny.
• Přidáme 400 µl vychlazeného 96% ethanolu (z mrazáku).
• Inkubujeme 15 min v termobloku při 37 °C.
• Centrifugujeme 5 min při 13 800 rpm.
• Supernatant opět opatrně slijeme do kádinky (už není nutné v digestoři).
• Přidáme 200 µl vychlazeného 70% ethanolu (z mrazáku) a necháme cca 5 min stát.
• Centrifugujeme 5 min při 13 800 rpm.
• Supernatant opět opatrně slijeme do kádinky a eppendorfky necháme cca 10 - 15 min stát a vyschnout.
Izolace DNA
4
• Přibližně 1–2 min vysušíme pelet v otevřených eppendorfkách na termobloku při 37 °C. Sušení ukončíme ve chvíli, kdy lze pelet poklepem (cvrnknutím) v uzavřené eppendorfce oddělit od stěny.
• Vysušený pelet rozpustíme ve 200 µl TE pufru nebo sterilní vody.
• Uzavřené eppendorfky inkubujeme 30 min na termobloku při 37 °C pro úplné rozpuštění.
• Krátce promícháme na vortexu a krátce stočíme na stolní centrifuze.
• DNA uchováváme v ledničce nebo dlouhodobě při –20°C (–80°C).
C) Extrakce DNA pomocí NaOH
Extrakce DNA pomocí NaOH je velmi levnou a rychlou metodou přípravy DNA. Hojně se používaná pro extrakci DNA z mechů.
Princip
Hydroxid sodný naruší buněčné stěny, DNA se uvolní do roztoku a stává se denaturovanou, řetězce jsou separované.
Postup
Obecné
• Takto získanou DNA obvykle není možné dlouhodobě skladovat (degraduje). Lze ji použít pro běžné metody založené na PCR. Pro některé aplikace náročné na kvalitu DNA (např. AFLP) ji použít nelze.
Izolace
• Část nebo celou rostlinku mechu vložíme do eppendorfky, uzavřeme a vymrazíme v tekutém dusíku. Vymrazíme homogenizátorek a krouživými pohyby drtíme vzorek na jemný prášek.
• K rozdrcenému materiálu přidáme 5 µl 0,5M NaOH a drtíme vzorek v roztoku.
• Přidáme dalších 15 µl 0,5M NaOH a pokračujeme v drcení další 1−2 min.
• Centrifugujeme 2 min 13 800 rpm.
• Supernatant přepipetujeme do nové eppendorfky a ředíme 1:10 roztokem 100mM Tris-HCl, pH 8,3. Podle potřeby dál ředíme roztokem 100mM Tris-HCl pufru.
• Skladujeme v mrazáku při –20°C max. 1 měsíc.
Izolace DNA
5
pip
pož
pož
puv
V
Vzř
C
C==
D) Měření koncentrace DNA a ředění DNA
Měření koncentrace DNA
• koncentraci DNA měříme pomocí spektrofotometru Biowave II (Biochrom)
• zaznamenáme: - koncentraci DNA v ng/µl - poměr A260/A280 – udává znečištění proteiny, u čisté DNA je 1,8 až 2,0 - poměr A260/A230 – pokud je méně než 1,8, značí to přítomnost organických
kontaminant – (poly)fenoly, thiokyanáty, sacharidy, močovina apod.; u čisté DNA se
pohybuje v rozmezí 1,8−2,0(–2,2)
Ředění DNA
• ředění vypočteme např. podle vzorečku:
Cpův ............... koncentrace původní (originálního vzorku) Cpož ............... koncentrace požadovaná (na kterou chci ředit) Vpož ............... objem požadovaný Vpip ............... objem pipetovaný (vzorku) zř .................. zředění
• Např. chceme 100 µl DNA o koncentraci 30 ng a máme k dispozici DNA o koncentraci 215 ng/µl.
Smícháme tedy 13,95 µl koncentrované DNA (našeho původního originálního vzorku) a 86.05 µl sterilní Milli-Q vody, tj. Vpož – Vpip (100 µl – 13,95 µl).
E) Otestování kvality DNA na agarosovém gelu
• připravíme 0,8 % agarosový gel
• cca 5 µl nenaředěné DNA smícháme se 2 µl nanášecího pufru LB (loading buffer) a naneseme na gel
• jako žebříček naneseme 6 µl Lambda/HindIII firmy NEB
95,1317,7
10010017,7
30
215==⇒== pip
pip
VV
PCR
6
Polymerázová řetězová reakce
PCR (Polymerase Chain Reaction)
Princip
Polymerázová řetězová reakce byla objevena v roce 1983 Kary Mullisem (Nobelova cena za chemii). Jde o jednoduchou metodu zmnožení (amplifikace) určité části DNA in vitro.
Základní uspořádání PCR vyžaduje vytvoření směsi reagencií a následné cyklické střídání teplot této směsi.
Složky PCR (PCR mix)
• templátová DNA – naše studovaná DNA nebo její část, kterou chceme namnožit (gen, část genu nebo nekódující sekvence)
• DNA polymeráza – enzym, který buduje nový řetězec DNA podle vzoru templátu
• PCR pufr – udržuje stabilní pH a obsahuje chemikálie pro optimální aktivitu a stabilitu DNA polymerázy
• MgCl2 – Mg2+ ionty slouží jako kofaktor DNA polymerázy
• nukleotidy (dNTP) – směs deoxynukleosid trifosfátů (dATP, dCTP, dGTP, dTTP), základních stavebních jednotek DNA
• primery – oligonukleotidy (krátké úseky jednořetězcové DNA), zpravidla o délce 10–25 bazí párující se s komplementární sekvencí v templátové DNA a sloužící jako počáteční místo replikace DNA, bez kterého by DNA polymeráza nemohla začít syntézu nového řetězce; k extenzi primeru dochází ve směru 5'→3' nově vznikajícího vlákna.
PCR teplotní program
Cyklické střídání teplot umožňuje přístroj zvaný termocykler.
• Na počátek cyklu se zařazuje počáteční denaturace (94–98°C, 1−10 min); následuje
• vlastní cyklus: - denaturace (94−98 °C, 20 s–1 min): dvoušroubovice DNA se rozplétá na dva řetězce
Pozn.: Pro GC bohaté DNA templáty může být denaturace prodloužena na 3-4 min.
- nasednutí primerů (annealing) (45−60°C, cca 20−45 s): primery se specificky párují s komplementární oblastí na templátové DNA Teplota nasedání primerů by měla být o 5°C nižší než teplota tání (denaturace), tzv. melting temperature (Tm) primerů. Pro primery kratší než 25 bp se Tm vypočítá podle rovnice: Tm = 4(G+C) + 2(A+T)
- syntéza DNA (elongace, extenze) (většinou 72 °C, 1 min) – optimální teplota pro činnost DNA polymerázy; na tvorbu 1 000 bazí je potřeba 1 min
• Finálním krokem je :
- konečná elongace (72°C, 5−15 min): dokončení syntézy částečných produktů
- zchlazení PCR reakce (4−15°C) Střídáním tří teplot dochází k exponenciálnímu množení požadovaného úseku DNA vymezeného dvojicí použitých primerů. Počet cyklů u běžné PCR se pohybuje od 25 do 40. Výsledkem PCR je pak 225-240 kopií příslušného úseku DNA z každé molekuly templátové DNA. DNA lze vizualizovat po inkorporaci UV fluorescenčního barviva (např. Syber Green, ethidiumbromid ) do dvoušroubovice DNA pod UV světlem.
PCR
7
Optimalizace PCR
Cílem optimalizace je specifický průběh PCR reakce, tj. aby vznikal pouze jediný produkt odpovídající amplifikovanému úseku. Výsledek PCR reakce proto ověřujeme na gelu. V případě, že produkt PCR reakce není zřetelný nebo je výsledkem více produktů odlišné délky (nespecifické produkty), reakce neproběhla optimálně. Je nutné změnit reakční podmínky tak, aby PCR proběhla úspěšně. Je několik možností:
• změna koncentrace DNA – snížením koncentrace DNA snížíme i koncentraci případných inhibitorů PCR, které mohou být přítomny ve vzorku, PCR může úspěšně proběhnout i s množstvím DNA menším než 1 ng; někdy naopak pomůže zvýšit koncentraci DNA až na 50 ng
• změna teploty nasednutí primeru (annealingu) – snížením umožníme lepší vazbu primerů na templátovou DNA, přílišné snížení však může vést k tvorbě nespecifických produktů. Ideální je provést gradientový pokus a následně vybrat nejvhodnější teplotu.
• zvýšení koncentrace MgCl2 – hořčík stabilizuje komplex DNA–polymeráza, zvýšení koncentrace na 2.5 – 6.0 mM (standardní koncentrace je 1.5-2.0 mM) může přinést úspěch.
• přidání aditiv – některé látky (DMSO, formamid, betaine, glycerol, BSA, (NH4)2SO4) mohou zvyšovat stabilitu polymerázy, specifičnost vazby primerů.
• modifikace cyklu – prodloužení (zkrácení) doby elongace, zvýšení počtu cyklů, apod.
• touch-down protokol – do teplotního programu se na počátek amplifikace zařadí 3–5 cyklů s vyšší teplotou nasedání primerů. V prvních cyklech vzniká určité minimum jen specifických produktů, tím se redukuje nespecifické nasedání primeru a snižuje se tvorba nespecifických produktů. V dalších cyklech se použije nižší teplota, která zajistí dostatečný výtěžek.
• úprava tzv. ramping time (rychlosti přechodu z jedné teploty na druhou) – např. AFLP vyžaduje pomalé ohřívání vzorku (při rychlém zahřívání část primerů odpadne), pro dostatečný výtěžek je nutné snížit ramping time na 1°C/s (standardně bývá 3°C/s).
PCR protokol s využitím Plain PP Master Mixu (Top-Bio)
Plain PP Master Mix již obsahuje PCR pufr, hořčík, nukleotidy a DNA polymerázu. Jeho použitím se výrazně snižuje čas nutný pro přípravu PCR reakce. Snížením počtu pipetovacích kroků je eliminována chybovost při přípravě.
Postup
• Po celou dobu pracujeme na ledu. Všechny potřebné reagencie necháme rozmrznout. Mezitím si označíme PCR zkumavky nebo stripy.
• Do 1.5 ml eppendorfky připravíme PCR směs pro příslušný počet vzorků + 1 vzorek (rezerva pro pipetovací chybu). Např. máme 10 vzorků, PCR směs připravíme pro 11 vzorků.
• Jednotlivé složky pipetujeme v pořadí: voda, primery a Plain PP Master Mix. Promícháme na vortexu a krátce stočíme na stolní centrifuze.
PCR
8
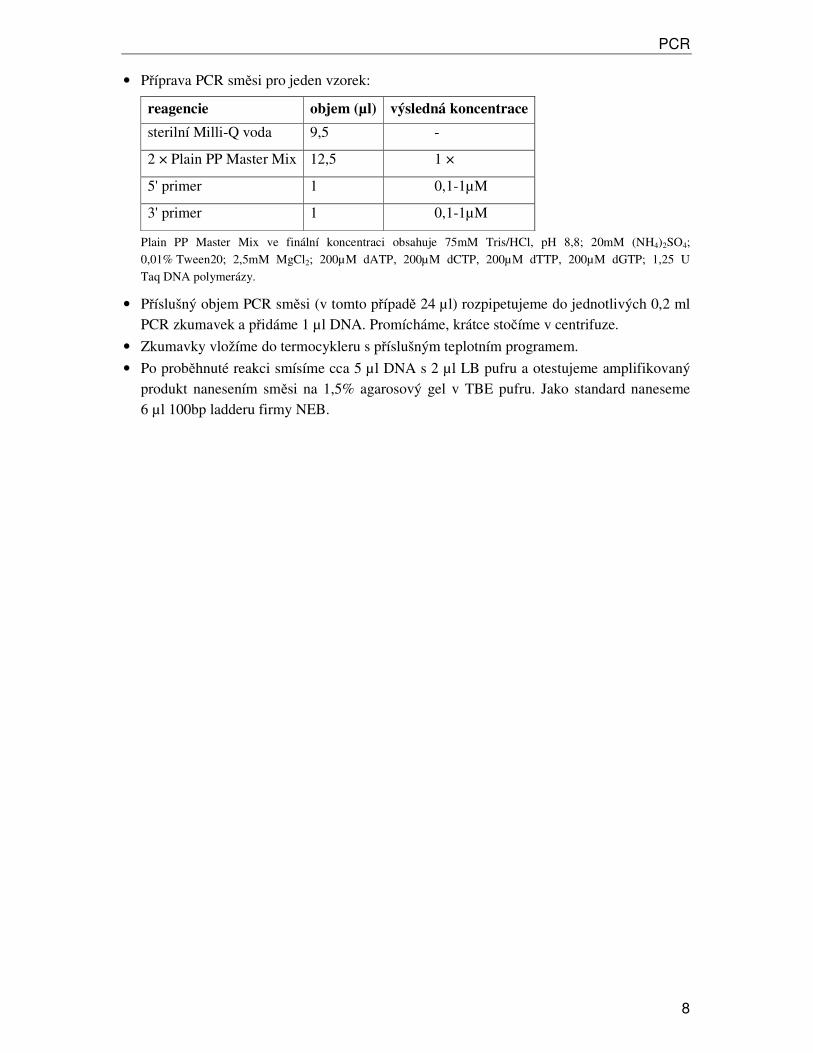
• Příprava PCR směsi pro jeden vzorek:
reagencie objem (µl) výsledná koncentrace
sterilní Milli-Q voda 9,5 -
2 × Plain PP Master Mix 12,5 1 ×
5' primer 1 0,1-1µM
3' primer 1 0,1-1µM
Plain PP Master Mix ve finální koncentraci obsahuje 75mM Tris/HCl, pH 8,8; 20mM (NH4)2SO4; 0,01% Tween20; 2,5mM MgCl2; 200µM dATP, 200µM dCTP, 200µM dTTP, 200µM dGTP; 1,25 U Taq DNA polymerázy.
• Příslušný objem PCR směsi (v tomto případě 24 µl) rozpipetujeme do jednotlivých 0,2 ml PCR zkumavek a přidáme 1 µl DNA. Promícháme, krátce stočíme v centrifuze.
• Zkumavky vložíme do termocykleru s příslušným teplotním programem.
• Po proběhnuté reakci smísíme cca 5 µl DNA s 2 µl LB pufru a otestujeme amplifikovaný produkt nanesením směsi na 1,5% agarosový gel v TBE pufru. Jako standard naneseme 6 µl 100bp ladderu firmy NEB.
Sekvenování
9
Sekvenování DNA
Princip
Sekvenováním zjistíme pořadí bazí (A,C,T,G) v konkrétním úseku DNA. Postup můžeme rozdělit do několika kroků: 1. PCR. Pomocí dvojice primerů amplifikujeme vybraný úsek DNA. Primery jsou specifické pro jedno konkrétní místo v jaderné nebo chloroplastové DNA. V jaderné (nukleární DNA, nDNA) se nejjednodušeji a nejčastěji sekvenují oblasti tzv. internal transcribed spaceru (ITS), které jsou přítomny až v desítkách tisíc kopií. Úseky chloroplastového genomu se také velmi jednoduše amplifikují, protože kruhových molekul cpDNA je v izolátu zpravidla velké množství. Často se sekvenují nekódující úseky (introny nebo spacery) jako např. trnL-trnF, psbA-trnH atd. 2. Přečištění (purifikace) PCR produktu. Zbylé primery a neinkorporované nukleotidy (dNTP) odstraníme pomocí komerčního kitu. To je velmi důležité, protože při následné sekvenační reakci je použit pouze jeden primer a také koncentrace dNTP musí být vybalancována vzhledem k ostatním složkám, především fluorescenčně značeným nukleotidům (ddNTP). 3. Cyklické sekvenování (cycle sequencing). Modifikovaná PCR reakce, při které použijeme pouze jeden sekvenační primer a PCR produkt jako templát. Nedochází tedy k exponenciální, ale pouze k lineární amplifikaci templátu. Kromě klasických dNTP jsou v sekvenační směsi přítomny i ddNTP (2´,3´-dideoxynukleotidtrisfosfáty). Ty jsou fluorescenčně značeny (každá baze jinou barvou) a při inkorporaci do vznikajícího řetězce DNA zamezí jeho dalšímu prodlužování. Výsledkem sekvenační reakce je tak směs různě dlouhých řetězců DNA a každý je fluorescenčně označen barvou podle toho, kterou bazí končí. Tyto produkty jsou pak elektroforeticky rozděleny na automatickém sekvenátoru a je možno přečíst posloupnost jednotlivých bazí. Sekvenuje se vždy jen s jedním primerem. Sekvenátor umožní číst cca 700 bazí, záleží na kvalitě PCR produktu a na sekvenci bazí. Pokud chceme sekvenovat delší úsek než cca 700 bazí, je třeba provést dvě sekvenační reakce s různými primery, s tzv. forward a reverse primerem. PCR produkt bude osekvenován z obou stran. Získané sekvence se potom spojí dohromady pomocí speciálního programu. 4. Purifikace produktů sekvenační reakce. Před analýzou na automatickém sekvenátoru je třeba produkty cyklického sekvenování přečistit, např. na Sephadexu. 5. Sekvenační analýza na automatickém sekvenátoru. V kapilárním sekvenátoru ABI PRISM 3130xl firmy Applied Biosystems jsou produkty cyklického sekvenování elektroforeticky rozděleny podle délky.
Sekvenování
10
Protokol na PCR amplifikaci
Viz obecný protokol k PCR.
Přečištění PCR produktu pomocí JETquick kitu (GENOMED)
PCR produkt přečistíme pomocí kitu GENOMED Jetquick PCR Purification Spin Kit, který využívá tzv. „spin column technique“, tj. kolonky s membránou, na kterou se váže DNA. Alternativou je QIAGEN QIAquick PCR Purification Kit, případně můžeme přečistit fragment vyřízlý z gelu pomocí QIAquick Gel Extraction Kit.
Obecné
• Před započetím práce zkontrolujeme přidání ethanolu do pufru H2 (zaškrtnutí políčka na lahvičce).
• Do 1,5 ml eppendorfky odpipetujeme potřebný objem elučního pufru a předehřejeme na 60–70°C. v termobloku. Předehřátí pufru je důležité v případě eluce PCR produktu delšího než 5 kb.
Postup
• Ke zbylým 20 µl PCR produktu přidáme 80 µl roztoku H1 a důkladně promícháme na vortexu. Pozn.:V případě jiného objemu PCR reakce přepočteme množství H1 roztoku, aby zůstal poměr zachován.
• Vložíme kolonku do označené 2 ml eppendorfky bez víčka (z kitu), přepipetujeme do ní směs z předchozího bodu.
• Centrifugujeme 1 minutu při 13 000 rpm.
• Odstraníme filtrát (to, co proteklo kolonkou), DNA zůstala vázána na membráně.
• Vložíme kolonku zpět do 2 ml eppendorfky a přidáme 500 µl roztoku H2.
• Centrifugujeme 1 minutu při 13 000 rpm.
• Opět odstraníme filtrát (DNA vázaná na membráně je promyta).
• Vložíme kolonku zpět do 2 ml eppendorfky a centrifugujeme 1 minutu při maximálních otáčkách (13 800 rpm), abychom důkladně odstranili roztok H2 z kolonky.
• Vložíme kolonku do nové (a označené!) 1,5 ml eppendorfky (není součástí kitu) a přímo těsně nad střed membrány pipetujeme 30–75 µl TE pufru (příp. sterilní vody) předehřátého na 60–70°C.
• 5 min inkubujeme na stole.
• Centrifugujeme 2 min při 13 000 rpm (DNA je eluována).
• DNA dále skladujeme při –20 °C .
Příprava vzorků pro sekvenování DNA
• Pro optimální průběh sekvenační reakce je potřeba zvolit správný poměr koncentrace primeru ku PCR produktu.
• Změříme koncentraci DNA pomocí spektrofotometru Biowave II.
• Pracujeme na ledu a v Biohazard boxu.
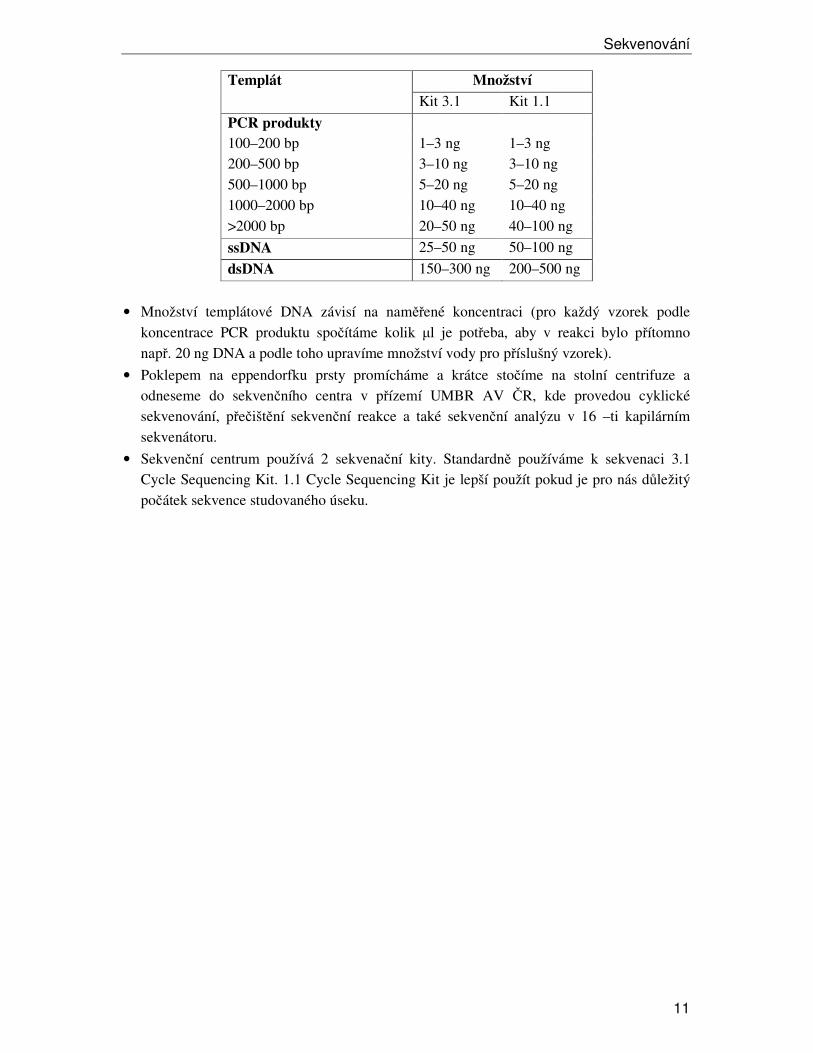
• Do 0,2 ml eppendorfek určených pro sekvenaci pipetujeme přečištěnou DNA v množství dle délky PCR produktu (viz tabulka), 3–5 pmol primeru a sterilní vodu do celkového objemu 15 µl.
Sekvenování
11
Množství Templát
Kit 3.1 Kit 1.1
PCR produkty 100–200 bp 1–3 ng 1–3 ng 200–500 bp 3–10 ng 3–10 ng 500–1000 bp 5–20 ng 5–20 ng 1000–2000 bp 10–40 ng 10–40 ng >2000 bp 20–50 ng 40–100 ng
ssDNA 25–50 ng 50–100 ng
dsDNA 150–300 ng 200–500 ng
• Množství templátové DNA závisí na naměřené koncentraci (pro každý vzorek podle koncentrace PCR produktu spočítáme kolik µl je potřeba, aby v reakci bylo přítomno např. 20 ng DNA a podle toho upravíme množství vody pro příslušný vzorek).
• Poklepem na eppendorfku prsty promícháme a krátce stočíme na stolní centrifuze a odneseme do sekvenčního centra v přízemí UMBR AV ČR, kde provedou cyklické sekvenování, přečištění sekvenční reakce a také sekvenční analýzu v 16 –ti kapilárním sekvenátoru.
• Sekvenční centrum používá 2 sekvenační kity. Standardně používáme k sekvenaci 3.1 Cycle Sequencing Kit. 1.1 Cycle Sequencing Kit je lepší použít pokud je pro nás důležitý počátek sekvence studovaného úseku.
ISSR
12
ISSR, Inter simple sequence repeat
Princip
Jedná se o metodu založenou na PCR, která využívá mikrosatelitové sekvence jako primery (16-25 bp). Mikrosatelity (SSR) jsou jednoduché krátké opakující se sekvence několika
nukleotidů (1−4 báze), tzv. repetice. Tyto repetitivní jednotky se vyskytují často, náhodně,
v celém genomu u všech eukaryot a liší se počtem opakování. Během PCR dochází k amplifikaci úseků DNA mezi dvěmi stejnými mikrosatelitovými repetitivními sekvencemi, které jsou umístěny v řetězci DNA v opačném směru. Výsledkem jsou různě dlouhé úseky DNA mezi mikrosatelity.
Technické provedení této metody je podobné jako u RAPD analýzy (RAPD – random amplified polymorphic DNA; metody sledující délku fragmentů DNA amplifikovaných pomocí určitého krátkého náhodně zvoleného primeru), kdy jsou využity tzv. arbitrární primery. Jde tedy o metodu univerzální, nevyžadující žádné znalosti o genomu zkoumaného organismu. ISSR nachází využití při studiu genetické diverzity, fylogeneze, mapování genomu a v evoluční biologii.
ISSR primery nesou sekvenční motiv komplementární k sekvenci mikrosatelitů.
Na jejich konci bývá často připojena „kotva“, tzv. anchor motiv. Nejčastěji 1−3 báze odlišné
od repetitivní mikrosatelitní sekvence, které při PCR amplifikaci zajistí specifické nasednutí na konec komplementárního templátového mikrosatelitního motivu.
Vlastní postup zahrnuje běžnou PCR za využití obvykle jednoho primeru. Méně často se používá kombinace dvou ISSR primerů. Produkty PCR amplifikace jsou vizualizovány na běžném agarózovém gelu. Výsledná sada vzniklých fragmentů odráží variabilitu v distribuci a délce mikrosatelitů v genomu. Analýzu každého vzorku je nutné provést ve dvou nezávislých PCR a do statistického hodnocení zahrnout pouze „proužky“, které se objeví v obou těchto nezávislých opakováních.
ISSR
13
Postup
PCR amplifikace
• Připravíme PCR směs pro příslušný počet vzorků + 1 rezerva jako negativní kontrola. Pracujeme stále na ledu. Pro 1 reakci o objemu 20 µl je složení následující:
• Promícháme, krátce centrifugujeme a pipetujeme po 19 µl do PCR stripů. Přidáme po 1 µl DNA vzorku. Promícháme poklepnutím prstem na strip a krátce centrifugujeme na stolní centrifuze.
• Vložíme do termocykleru a spustíme odpovídající teplotní program s použitím tzv. touchdown protokolu.
Vizualizace na agarosovém gelu
• Připravíme 1,8 % agarózový gel.
• Po skončení programu stripy vyndáme z cykleru a krátce centrifugujeme.
• Smícháme 1 µl LB s 3 µl PCR produktu, promícháme a naneseme na gel.
• Jako žebříček použijeme 4 µl 100 bp Ladderu (před a za vzorky).
• Napětí nastavíme na 40 V, po vyjetí vzorků z jamek zvýšíme na 80 V, elektroforézu necháme běžet cca 4 h.
• Pořizujeme dvě fotky gelu, první na obvyklou expozici a druhou přeexponovanou (vyniknou i slabší, tj. méně amplifikované produkty).
Mikrosatelity
14
Analýza mikrosatelitů
Princip
Mikrosatelitní DNA neboli mikrosatelity, také nazývané STR (short tandem repeats – krátká tandemová opakování) nebo SSR (simple sequence repeats), jsou krátké segmenty DNA, ve kterých se mnohokrát opakují specifické motivy nukleotidových sekvencí. Nejčastěji jsou tvořeny opakováním mono-, di-, tri- nebo tetranukleotidů, např. (A)n, (AT)n, (ATA)n
apod.
Počet opakování jednotky (repetice) v konkrétním místě DNA (lokusu) definuje alelu. Každý jedinec má v jaderné DNA dvě kopie každého mikrosatelitu, jeden zděděný po matce, druhý po otci. U diploidních jedinců tak můžeme odhalit dvě alely, u tetraploidních až čtyři atd. Délku alel(y) zjistíme PCR amplifikací daného lokusu pomocí primerů přiléhajících k mikrosatelitní sekvenci. Pro analýzu mikrosatelitů tedy potřebujeme znát okolní sekvence. PCR fragmenty pak rozdělíme podle délky v automatickém sekvenátoru tzv. fragmentační analýzou. Ke každému vzorku se přidá fluorescenčně značený standard (500-LIZ), obsahující fragmenty DNA o známé délce, aby bylo později možné identifikovat délku každého fragmentu (alely) a jednoznačně ji srovnávat s délkou fragmentů v jakémkoliv jiném běhu.
Mikrosatelity jsou považovány za jedny z nejvhodnějších genetických markerů díky jejich hojnému výskytu v celém genomu, extrémní variabilitě (polymorfismu) a kodominantní dědičnosti (umožňuje rozlišit heterozygoty). V populační biologii nacházejí využití při identifikaci příbuzných jedinců, až po odvozování demografických parametrů.
Jaderné mikrosatelity jsou často velmi druhově specifické, musíme tedy pracovat s druhem, pro nějž jsou primery již publikované. Používají se na vnitrodruhové úrovni. Chloroplastové mikrosatelity se naopak vyskytují v místech mezi konzervovanými sekvencemi. Na základě tzv. konsenzuálních primerů můžeme určité lokusy amplifikovat pro druhy z mnoha různých čeledí. Jsou tedy využitelné pro hodnocení variability mezi druhy.
Postup
1) PCR
Pro amplifikaci konkrétního úseku DNA, který obsahuje repetitivní mikrosatelitovou sekvenci použijeme dva specifické primery, jeden z primerů je fluorescenčně značený (používáme barvy 6-FAM, VIC, NED a PET).
Pokud je třeba analyzovat více mikrosatelitových lokusů (často se analyzuje 8−10), je
výhodné pokusit se sestavit tzv. multiplex PCR. Je to v podstatě běžná PCR reakce, do které přidáme více primerových páru najednou a amplifikujeme tak více lokusů (až třeba pět) v jedné PCR reakci. Můžeme tak výrazně snížit finanční i časové náklady na analýzu. Pro úspěšnou a dostatečnou amplifikaci všech lokusů musíme zaručit, aby (1) primery měly přibližně stejnou teplotu annealingu, (2) primery v jedné PCR reakci nevytvářely dimery, (3) lokusy s překrývajícím se rozsahem předpokládaných délek fragmentů byly označeny jinou fluorescenční barvou.
Mikrosatelity
15
• Do 1.5 ml eppendorfky napipetujeme směs pro příslušný počet vzorků + 1. Pracujeme stále na ledu.
• Promícháme na vortexu, krátce stočíme v centrifuze a po 14 µl pipetujeme do PCR zkumavek (případně stripů).
• Přidáme 1 µl naředěné DNA (5 ng/µl), promícháme na vortexu a vložíme do termocykleru s příslušným teplotním programem.
• Po proběhnutí reakce otestujeme úspěšnost amplifikace na 1% agarosovém gelu v TBE pufru.
Příprava pro fragmentační analýzu
• Připravíme směs osahující 0,25 µl velikostního standardu (Size standard, 500-LIZ), 0,5 µl od každého PCR produktu a doplníme deionizovaným formamidem (tzv. Hi-Di formamid = high deionized) na výsledný objem 10 µl.
• Postupujeme tak, že si nejprve připravíme směs formamidu a velikostního standardu pro příslušný počet vzorků +1.
• Promícháme, krátce stočíme, a rozpipetujeme do označených 0,5 ml eppendorfek.
• Přidáme PCR produkt(y).
• Promícháme na vortexu, krátce stočíme v centrifuze a odneseme do sekvenačního centra.
PCR-RFLP
16
PCR-RFLP (Restriction Fragment Lenght Polymorphism)
Princip
Metoda využívá tzv. restrikčních endonukleas, které specificky štěpí řetězec DNA. Např. enzym EcoRI rozštěpí DNA vždy, pokud nalezne sekvenci 3´-GAATTC-5´. Vlastní postup se skládá z PCR amplifikace určitého úseku DNA a jeho štěpení vybranou restriktasou. Restrikční fragmenty dělíme elektroforeticky podle velikosti. Pokud PCR produkt obsahuje právě jedno štěpné místo, získáme dva kratší fragmenty nebo více fragmentů, pokud je štěpných míst více. Restrikční místa mohou mutacemi zanikat i vznikat. Hledáme polymorfismus ve štěpných místech a ten pak interpretujeme jako genetickou podobnost studovaných organismů.
Postup
1) PCR amplifikace
• Viz obecný protokol pro PCR.
• Výsledek PCR ověřujeme standardním způsobem. Pro PCR-RFLP je důležité, aby byl na gelu pouze jeden specifický pruh.
2) Restrikce
Každá restriktáza má svůj specifický restrikční pufr, ve kterém vykazuje maximální aktivitu. Restrikční pufry jsou dodávány s příslušným enzymem, např. od firmy Fermentas, Takara nebo NEB (plakát na zdi vedle mrazáku) a zajišťují stálé optimální pH během restrikce. Stálou optimální teplotu pro restrikční reakci pak zajistíme v termostatu nebo termocykleru.
• Pracujeme stále na ledu.
• Do sterilní 1,5 ml eppendorfky připravíme restrikční směs pro příslušný počet vzorků + 1.
• Promícháme, krátce stočíme na stolní centrifuze a rozpipetujeme do jednotlivých 0,2ml PCR zkumavek po 12,4 µl. Přidáme 4 µl amplifikačního produktu, promísíme, stočíme a zkumavky vložíme na 4−5 h nebo přes noc do termocykleru nastaveného na teplotu 37 °C.
• Po skončení restrikce přidáme 3 µl nanášecího pufru LB (barvička zastaví restrikční reakci) a naneseme na 1,7% agarosový gel v TBE pufru spolu se 6 µl 100 bp žebříčku. Pro větší rozlišení lze použít vertikální polyakrylamidový gel.
AFLP
17
AFLP (Amplified Fragment Length Polymorphism)
Princip
Jde o restrikční metodu, která umožňuje analýzu polymorfismu v celkové genomové DNA a nevyžaduje znalosti o struktuře geonomu (specifické primery apod.). DNA je štěpena dvěma různými restrikčními enzymy a z velkého množství vzniklých fragmentů je pomocí dvou následných PCR selektována část fragmentů. (desítky až stovky). Sleduje se přítomnost nebo nepřítomnost fragmentu určité délky. Prvním krokem AFLP metody je restrikce. Pro štěpení DNA se používají restriktasy MseI a EcoRI, které štěpí DNA řetězec tzv. s přesahem, tj. na koncích fragmentů vznikají jenovláknové přesahy několika bazí. Na tyto volné konce se komplementárně vážou tzv. adaptory, krátké syntetické úseky dvouřetězcové DNA o známé sekvenci. Spojení adaptorů s konci restrikčních fragmentů zajišťuje enzym T4 DNA ligasa. Následuje tzv. preselektivní amplifikace, kdy PCR reakce probíhá s dvojicí primerů, které mají sekvenci komplementární k sekvenci adaptorů a obsahují jednu tzv. selektivní bázi navíc (obecně se používá C pro MseI primer a A pro EcoRI primer). Výsledkem PCR jsou fragmenty s MseI adaptorem na jednom konci a EcoRI adaptorem na druhém konci. Díky prodloužení o jednu selektivní bázi vzniká pouze 1/16 restrikčních fragmentů. Preselektované fragmenty vstupují do druhé, tzv. selektivní PCR, ve které jsou použity primery komplementární k adaptorům prodloužené o tři selektivní báze (C + dvě náhodně zvolené báze pro MseI a A + dvě náhodně zvolené báze pro EcoRI). Tím je počet fragmentů znovu redukován. EcoRI primery jsou fluorescenčně značené, což umožňuje separaci fragmentů v automatickém sekvenátoru tzv. fragmentační analýzou. Fragmenty jsou rozděleny podle délky, jejíž přesné určení umožní přidání fluorescenčně značeného standardu obsahujícího fragmenty DNA o známé délce ke každému vzorku.
Obvykle se hledá několik primerových kombinací, které pak budou odlišeny různou fluorescenční barvou EcoRI primeru a mohou být na sekvenátoru analyzovány zároveň ve směsných vzorcích. Automatický sekvenátor ABI PRISM 3130xl firmy Applied Biosystems umožňuje současně analýzu čtyř různých primerových kombinací.
Díky možnosti detekovat variabilitu na nízkých úrovních lze AFLP použít pro studium procesů v populacích, odlišení blízkých taxonů, při fylogeografických studiích apod. Existují ale i práce využívající AFLP pro studium fylogeneze i na rodové úrovni. Nevýhodou metody je poměrně vysoká cena, časová náročnost metody i požadavky na kvalitu DNA a přesné reakční podmínky při restrikci i PCR.
V současnosti je možné v naší laboratoři použít dva protokoly:
AFLP
18
A) Protokol s využitím AFLP kitů od firmy Invitrogen
Obecné poznámky:
• Sekvenační centrum vyžaduje pro fragmentační analýzu, aby byl výsledný počet vzorků v násobcích 16.
• Během celého postupu pracujeme stále na ledu. Enzymy vyndáváme z mrazáku až těsně před jejich použitím a ihned poté vracíme zpět.
• Fluorescenčně značené primery chráníme před světlem (zkumavky obalujeme alobalem a necháváme rozmrzat např.v tmavé skříni). Pro práci používáme alikvoty.
1) Restrikce
• Využijeme AFLP® Core Reagent Kit I.
• Připravíme si restrikční směs pro patřičný počet vzorků + 1. Výsledný objem směsi je 2,5 µl na vzorek.
• Směs promícháme, krátce centrifugujeme a přidáme po 5 µl ke vzorkům po restrikci; celkový objem 10 µl.
• Vložíme do termocykleru a spustíme restrikci: 12 hodin při teplotě 37°. Pozn.: Nejlépe spustit odpoledne a nechat proběhnout přes noc, aby bylo možné ráno pokračovat preselektivní amplifikací.
• Po proběhnutí reakce otestujeme úspěšnost restrikce/ligace na 1,8 % agarosovém gelu v TBE pufru. Nanášíme 3–5 µl směsi po ligaci spolu se 3 µl LB a 3 µl 100 bp ladderu. Na gelu by měl být vidět „smír“ restrikčních fragmentů (zejména v oblasti 50–1000 bp).
3) Preselektivní amplifikace
• Využijeme AFLP® Pre-amp Primer Mix I .
• Připravíme si preamplifikační směs pro patřičný počet vzorků + 1. Výsledný objem směsi je 5 µl.
PA mix ........................................ 4 µl PCR buffer (10×)......................... 0,5 µl DNA polymerasa (1 U/µl) ........... 0,1 µl
AFLP
19
• Směs promícháme, krátce centrifugujeme, a rozpipetujeme po 4,6 µl do PCR stripů nebo 0,2 ml PCR zkumavek.
• Přidáme 0,4 µl DNA po ligaci; výsledný objem 5 µl.
• Vložíme do termocykleru a spustíme program pro preselektivní amplifikaci:
1× 72°C 2 min 20× 94°C 1 s 56°C 30 s 72°C 2 min ramping 2°C/s 1× 60°C 30 min 1× 10°C hold
• Směs po preselektivní amplifikaci naředíme 10× naředíme (2 µl preamplifikační směsi + 18 µl sterilní H2O). Zbytek nenaředěného vzorku uschováme v –20°C pro případné opakování.
• Úspěšnost preselektivní amplifikace ověříme na 1,8 % agarosovém gelu v TBE pufru. Nanášíme 3 µl (neředěné) preamplifikační směsi spolu s 3 µl LB a 3 µl 100 bp ladderu. Na gelu by měl být vidět „smír“ preamplifikačních fragmentů, na několika místech intenzivnější.
4) Selektivní amplifikace
• Připravíme si mix pro odpovídající počet vzorků + 1. Obvykle děláme pro každý vzorek 4 varianty, které se liší použitými primery (neznačený MseI a fluorescenčně značený EcoRI). Výsledný objem směsi je 7,5 µl na jeden vzorek v jedné variantě.
• Směs promícháme, krátce centrifugujeme a rozpipetujeme po 7,5 µl do PCR stripů nebo 0,2 ml PCR zkumavek.
• Přidáme 2,5 µl ředěné DNA po preselektivní amplifikaci, výsledný objem směsi 10 µl.
• Promícháme, krátce centrifugujeme na stolní centrifuze.
• Vložíme do cykleru a spustíme program pro selektivní amplifikaci:
1× 94°C 2 min 65°C 30 s 72°C 2 min s ramping 2°C/s 8× 94°C 1 s 64-56°C 30 s touch-down by 1°C 72°C 2 min ramping 2°C/s 23× 72°C 1 s 56°C 30 s 72°C 2 min ramping 2°C/s 1× 60°C 30 min 1× 10°C hold
AFLP
20
5) Příprava pro fragmentační analýzu
• Připravíme směs produktů selektivní amplifikace, které pocházejí z jednoho původního vzorku (jedné směsi po preamplifikaci), ale liší se fluorescenční barvou. Smícháme po 1 µl (nebo optimalizované objemy) od každé ze čtyř barev.
• Pokud není možné odnést vzorky na fragmentační analýzu v krátké době, přesrážíme selektivní PCR produkt octanem sodným (viz protokol C).
• Připravíme si mix pro patřičný počet vzorků + 1. Formamid je v laboratoři k dispozici v alikvotech pro přípravu 17 vzorků. Výsledný objem směsi je 10,5 µl na vzorek.
• Ke směsi přidáme 0,5 µl smíchané DNA po selektivní amplifikaci. Takto připravené vzorky ihned odneseme do sekvenančního centra na fragmentační analýzu.
B) Protokol bez využití AFLP kitů
• Obecné poznámky viz protokol A.
• ATP je skladováno při –80°C, proto potřebné množství necháme rozmrznout v předstihu.
1) Restrikce
• DNA naředíme TE pufrem tak, abychom měli 50 ng DNA v celkovém objemu 40 µl.
• Připravíme si restrikční směs pro patřičný počet vzorků + 1. Výsledný objem směsi je 10 µl na vzorek.
• Směs promícháme a rozpipetujeme po 10 µl do PCR stripů nebo 0,2 ml PCR zkumavek.
• Přidáme 40 µl DNA, výsledný objem 50 µl. Promícháme, krátce centrifugujeme na stolní centrifuze.
• Vložíme do cykleru a inkubujeme 16 hodin při teplotě 37°C Pozn.: Nejlépe spustit odpoledne a nechat proběhnout přes noc, aby bylo možné ráno pokračovat ligací.
2) Ligace
• Připravíme si ligační směs pro odpovídající počet vzorků + 1.
• Směs promícháme a rozpipetujeme po 10 µl do PCR stripů nebo 0,2 ml PCR zkumavek.
• Přidáme 50 µl DNA po restrikci. Výsledný objem ligační směsi je 60 µl.
• Promícháme, krátce centrifugujeme na stolní centrifuze.
• Vložíme do cykleru a spustíme program pro ligaci: 3 hodiny při 37°C.
• Po proběhnutí reakce otestujeme úspěšnost restrikce/ligace na 1,8 % agarosovém gelu v TBE pufru. Nanášíme 10 µl směsi po ligaci spolu se 3 µl LB a 3 µl 100 bp ladderu. Na gelu by měl být vidět „smír“ restrikčních fragmentů (zejména v oblasti 50–1000 bp).
• Směs promícháme a rozpipetujeme po 45 µl do PCR stripů nebo 0,2 ml PCR zkumavek.
• Přidáme 5 µl naředěné DNA po ligaci. Výsledný objem směsi je 50 µl.
• Promícháme, krátce centrifugujeme na stolní centrifuze.
• Vložíme do cykleru a spustíme program pro preselektivní amplifikaci:
1× 94°C 1 min 30× 94°C 30 s 60°C 30 s 72°C 60 s ramping 1°C/s 1× 72°C 9 min 1× 10°C hold
• Úspěšnost preselektivní amplifikace ověříme na 1,2 % agarosovém gelu v TBE pufru. Nanášíme 10 µl preamplifikační směsi spolu s 3 µl LB a 3 µl 100 bp ladderu. Na gelu by měl být vidět „smír“ preamplifikačních fragmentů, na několika místech intenzivnější.
• Směs po preselektivní amplifikaci 20× ředíme (5 µl preamplifikační směsi + 95 µl TE pufru). Zbytek nenaředěného vzorku uschováme v –20°C pro případné opakování.
AFLP
22
4) Selektivní amplifikace
• Připravíme si mix pro odpovídající počet vzorků + 1. Obvykle děláme pro každý vzorek 4 sady vzorků, které se liší použitými primery (neznačený MseI a fluorescenčně značený EcoRI). Výsledný objem směsi je 7,5 µl na jeden vzorek v jedné sadě.
• Směs promícháme, krátce centrifugujeme a rozpipetujeme po 7,5 µl do PCR stripů nebo 0,2 ml PCR zkumavek.
• Přidáme 2,5 µl ředěné DNA po preselektivní amplifikaci, výsledný objem směsi je 10 µl.
• Promícháme, krátce centrifugujeme na stolní centrifuze.
• Vložíme do cykleru a spustíme program pro selektivní amplifikaci:
1× 94°C 2 min 10× 94°C 30 s 65-56°C 30 s touch-down by 1°C 72°C 60 s ramping 1°C/s 25× 72°C 9 min 56°C 30 s 72°C 60 s ramping 1°C/s 1× 72°C 15 min 1× 10°C hold
5) Příprava pro fragmentační analýzu
• Připravíme směs produktů selektivní amplifikace, které pocházejí z jednoho původního vzorku (jedné směsi po preamplifikaci), ale liší fluorescenční barvou. Smícháme po 1 µl (nebo optimalizované objemy) od každé ze čtyř barev.
• Pokud není možné odnést vzorky na fragmentační analýzu v krátké době, přesrážíme selektivní PCR produkt octanem sodným (viz protokol C).
• Připravíme si mix pro patřičný počet vzorků + 1. Formamid je v laboratoři k dispozici v alikvotech pro přípravu 17 vzorků. Výsledný objem směsi je 10,5 µl na vzorek.
• Ke směsi přidáme 0,5 µl smíchané DNA po selektivní amplifikaci. Takto připravené vzorky ihned odneseme do sekvenančního centra na fragmentační analýzu.
AFLP
23
C) Přesrážení octanem sodným
Všechny centrifugační kroky probíhají při 4°C v předem vychlazené centrifuze.
• Na stěnu 1,5 ml eppendorfky pipetujeme 1 µl 3M octanu sodného.
• Do kapky octanu sodného přidáme 3 µl směsi DNA po selektivní amplifikaci.
• Přidáme 25 µl čistého 96% ethanolu.
• Promícháme na vortexu, krátce stočíme na stolní centrifuze.
• Necháme stát 20 min v mrazáku při –20°C.
• Centrifigujeme 30 min při 14 000 rpm. Eppendorfky v centrifuze orientujeme do stejné pozice, abychom měli DNA usazenou vždy na stejném místě.
• Pomalu a opatrně slijeme supernatant, DNA zůstává usazená na stěně eppendorfky.
• Přidáme 100 µl 70% ethanolu.
• Centrifigujeme 5 min při 14 000 rpm.
• Pomalu a opatrně slijeme supernatant, DNA zůstává na stěně eppendorfky.
• Otevřené eppendofky necháme stát ve stojánku 5 min při laboratorní teplotě a následně dosušíme 10 min v termobloku při 65°C.
• Skladujeme v lednici (4°C).
• Před přípravou pro fragmentační analýzu rozpustíme ve 3 µl sterilní destilované vody (nebo TE pufru).
Elektroforéza DNA
24
Elektroforéza DNA
Horizontální agarosová elektroforéza
• pracujeme v rukavicích, protože všechny věci mohou být kontaminovány SyberGreenem (potencionální mutagen!)
• do Erlenmeyerovy baňky na předvážkách navážíme příslušné množství agarosy (nejčastější navážky viz tabulka)
• vložíme na několik minut do mikrovlnky na 550-700W; v uvařeném gelu nesmí být viditelné krystalky („nitky“) agarosy
• mezitím si připravíme vaničku na nalití gelu – položíme ji na nalévací stolek (vodováhy v rozích)
• podle počtu nanášených vzorků vybereme hřeben s dostatečným počtem zubů a umístíme ho do zářezů ve vaně
• uvařený gel ochladíme kroužením „Erlenkou“ pod tekoucí vodou (používáme rukavici, abychom se nespálili) na teplotu cca 50°C (odhadneme tak, že udržíme ruku na dně baňky)
• gel pomalu naléváme do vaničky s hřebínky, špičkou odstraníme případné bublinky v gelu a necháme tuhnout cca 30 minut
• po ztuhnutí opatrně vyjmeme hřebínky, vaničku s gelem vložíme do elektroforetické vany a zalijeme pufrem (1×TBE) tak, aby celý gel byl těsně pod hladinou a všechny jamky byly zality
• krajní jamky necháváme prázdné, do druhé jamky pipetujeme cca 6 µl 100bp Ladderu (žebříčku) a do dalších jamek pak směs DNA s nanášecím pufrem LB (Loading Buffer)
• po nanesení všech vzorků na gel přiklopíme víko s přívodními kabely, připojíme elektroforézu ke zdroji; napětí nastavíme v modu „SET“ na 50 - 100 V (dle velikosti gelu) otáčením knoflíku
• pro spuštění elektroforézy vypneme mod „SET“; po vyjetí vzorků z jamek napětí zvýšíme
• ve chvíli, kdy čelo (nejrychlejší barvička) dojíždí ke konci gelu, vypneme proud, sejmeme víko elektroforetické aparatury, vyndáme gel a umístíme do komory UV transiluminátoru dokumentačního systému Gel Imager
Elektroforéza DNA
25
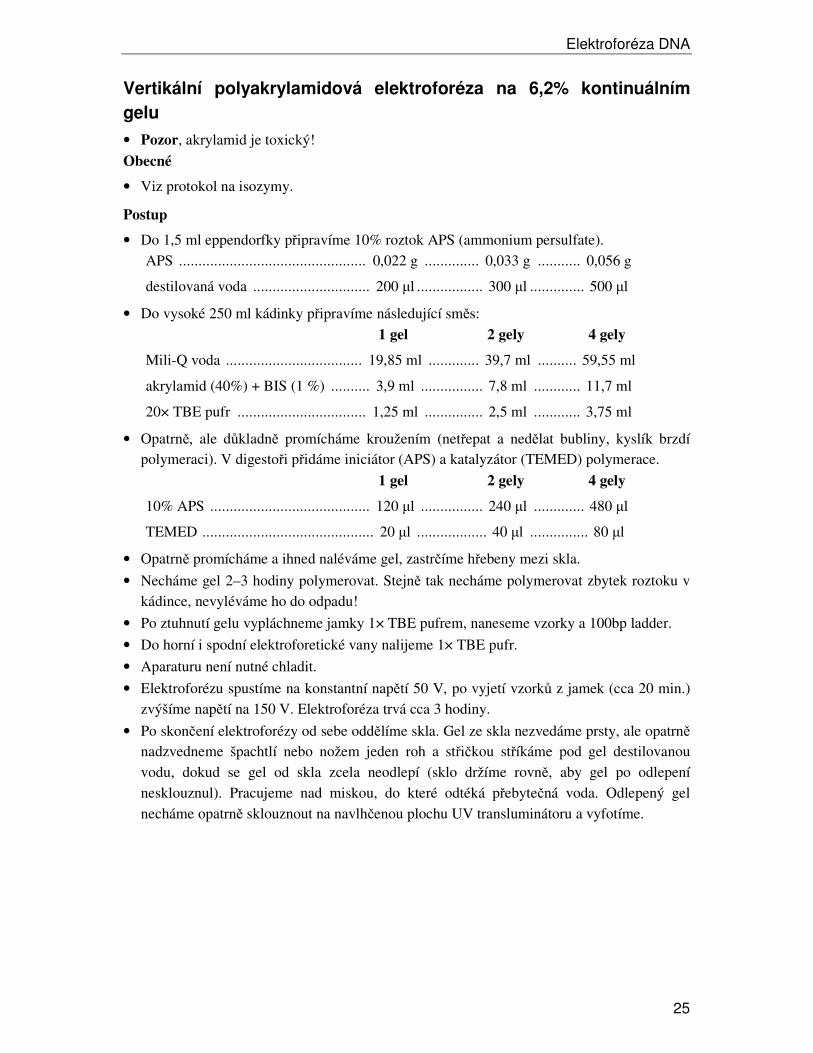
Vertikální polyakrylamidová elektroforéza na 6,2% kontinuálním
gelu
• Pozor, akrylamid je toxický!
Obecné
• Viz protokol na isozymy.
Postup
• Do 1,5 ml eppendorfky připravíme 10% roztok APS (ammonium persulfate). APS ................................................ 0,022 g .............. 0,033 g ........... 0,056 g
• Opatrně promícháme a ihned naléváme gel, zastrčíme hřebeny mezi skla.
• Necháme gel 2–3 hodiny polymerovat. Stejně tak necháme polymerovat zbytek roztoku v kádince, nevyléváme ho do odpadu!
• Po ztuhnutí gelu vypláchneme jamky 1× TBE pufrem, naneseme vzorky a 100bp ladder.
• Do horní i spodní elektroforetické vany nalijeme 1× TBE pufr.
• Aparaturu není nutné chladit.
• Elektroforézu spustíme na konstantní napětí 50 V, po vyjetí vzorků z jamek (cca 20 min.) zvýšíme napětí na 150 V. Elektroforéza trvá cca 3 hodiny.
• Po skončení elektroforézy od sebe oddělíme skla. Gel ze skla nezvedáme prsty, ale opatrně nadzvedneme špachtlí nebo nožem jeden roh a střičkou stříkáme pod gel destilovanou vodu, dokud se gel od skla zcela neodlepí (sklo držíme rovně, aby gel po odlepení nesklouznul). Pracujeme nad miskou, do které odtéká přebytečná voda. Odlepený gel necháme opatrně sklouznout na navlhčenou plochu UV transluminátoru a vyfotíme.
Elektroforéza DNA
26
Práce s dokumentačním systémem Gel Imager a softwarem Scion
VisiCapture
• Při manipulaci s gelem, nalévací vaničkou a elektroforetickou vanou pracujeme v rukavicích!
• Po skončení elektroforézy vypneme zdroj a odpojíme od něj kabely.
• Vyjmeme vaničku s gelem z elektroforetické vany.
• Do komory fotodokumentačního zařízení umístíme již samotný gel a otočíme ho o 90° (horní jamky jsou blíž levé straně komory transluminátoru), opatrným posouváním po ploše se zbavíme případných vzduchových bublin pod gelem, gel srovnáme.
• Bez rukavic (!) spustíme program Scion VisiCapture.
• Klikneme na „Image“ a vybereme „Start Live Capturing“.
• Při otevřených dvířkách komory si gel připravíme k focení, přímo na objektivu kamery můžeme vyladit zoom (velikost gelu nebo jeho části) a clonu (jas).
• Zavřeme dvířka a zapneme UV světlo, popřípadě ještě doladíme obraz (expozici lze vyladit kliknutím na „Image“ a „Properties“ – nastavením délky expozice nebo elektronického zesílení signálu, tj. gain).
• Klikneme na „Image“ a gel vyfotíme tlačítkem „Snap“.
• Obrázek uložíme vybráním „File“ a „Save As“ do příslušné složky .
• Vypneme zdroj UV světla, otevřeme dvířka, gel v rukavicích (!) zabalíme do papírového ručníku a vyhodíme do odpadkového koše k tomu určeného a očistíme plochu UV transiluminátoru ethanolem.
DNA žebříčky (ladders) používané v laboratoři (NEB)
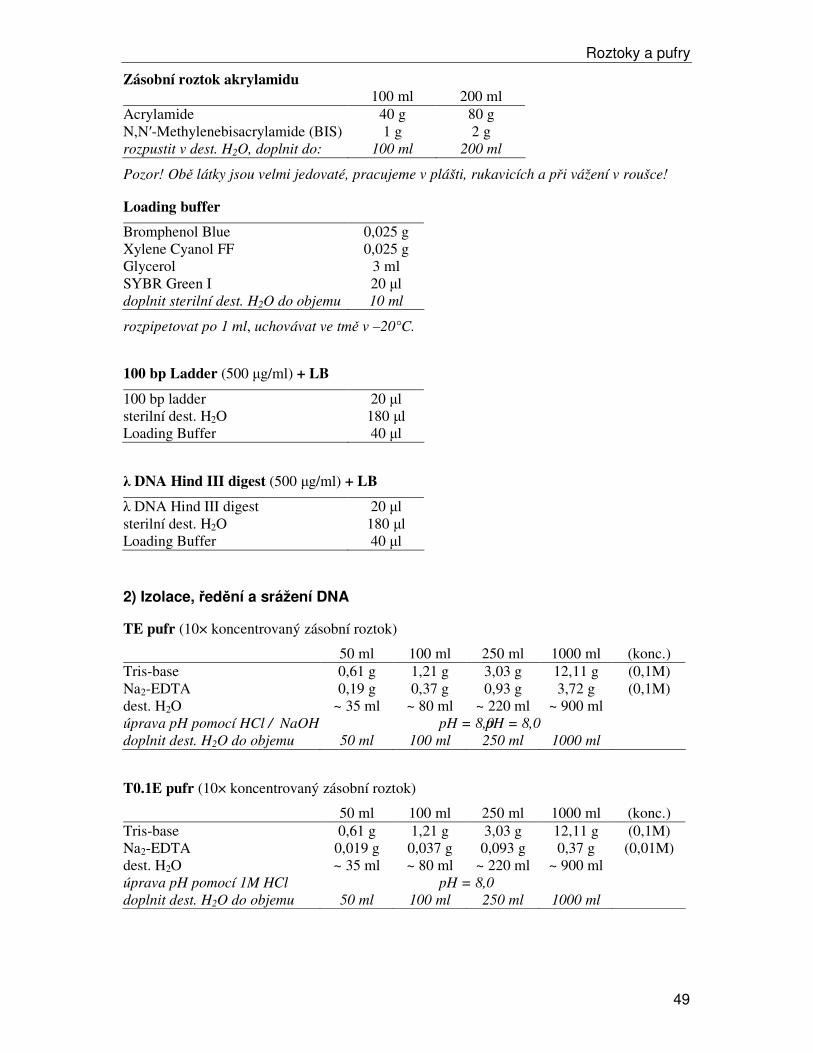
100 bp DNA Ladder (NEB)
0.5µg 100 bp DNA žebříčku/jamku,
1,3% agarosový gel v 1X TAE
Lambda DNA-Hind III Digest (NEB)
0.5 µg Lambda DNA-Hind III Digest/jamku,
1,0% agarosový gel
Isozymy
27
Isozymy (allozymy)
Princip metody
Analýza isozymů sleduje variabilitu na úrovni proteinů, konkrétně určitých enzymů, které jsme schopni specificky detekovat. Ačkoliv se v molekule proteinu díky degeneraci genetického kódu neprojeví všechny změny ve struktuře DNA, lze obvykle najít enzymy, které: (a) jsou u studovaných organismů v různých jedincích/populacích/taxonech zastoupeny různými formami, které se liší pohyblivostí při elektroforéze a (b) a tyto rozdíly jsou dědičné. Při kombinaci dat z několika enzymů dohromady můžeme získat dostatečnou variabilitu pro odhad obvyklých populačně-genetických parametrů, studium rozdílů mezi taxony a nebo dokonce identifikaci jedinců (isozymová analýza se u rostlin často využívá k identifikaci jednotlivých klonů).
Proteiny vyizolované z rostlinné tkáně naneseme na gel (škrobový nebo polyakrylamidový), rozdělíme elektroforézou a následně ze směsi detekujeme vybraný enzym (resp. skupinu enzymů, enzymový systém). Využíváme tzv. histochemické barvení, při kterém detekujeme projevy reakce katalyzované zkoumaným enzymem – na gelu dostáváme v místě aktivity enzymu barevnou zónu. Detekce je u různých enzymů různě specifická – někdy dostáváme na gelu jednu zónu, označovanou jako lokus, která odpovídá jednomu enzymu pravděpodobně kódovanému jedním genem, častěji ale získáváme více lokusů. Přítomnost více lokusů může být způsobena dvěma faktory – buď je daný enzym v genomu kódován několika geny nezávisle (např. jednou v jádře a jednou v plastidech) nebo není naše detekce zcela specifická a jedním barvením detekujeme více různých enzymů najednou. Právě v počtu lokusů je rozdíl mezi pojmy isozymy a allozymy. Jako isozymy (isoenzymy) se označují všechny různé formy enzymu (ve smyslu různé pohyblivosti při elektroforéze), které detekujeme jednou barvící reakcí. Jako allozymy se označují pouze různé formy v rámci jednoho lokusu – tedy různé alely jednoho genu. Pojem isozymy je obecnější, allozymy jsou podmnožinou isozymů.
V současnosti je známo více než 100 specifických barvení (i když asi v žádné laboratoři není možné dělat všechny; u nás máme v současnosti materiál pro 21 enzymových systémů, rutinně používáme 10–12 z nich). Většina používaných enzymů patří mezi enzymy základního metabolismu (např. glykolýza, pentosafosfátový cyklus, Krebsův cyklus, metabolismus aminokyselin). To znamená, že tytéž enzymy zastoupeny u široké škály organismů a isozymová analýza je v tomto ohledu dosti univerzální metodou. Za druhé jsou enzymy (resp. geny je kódující) pod velkým selekčním tlakem – enzymy pro základní metabolismus musí v organismu fungovat. Proto lze předpokládat, že případné různé formy enzymů (tedy isozymy/allozymy) budou fungovat přibližně stejně, rozdíly budou ± selekčně neutrální. To je ideální stav pro výpočet populačně-genetických parametrů. (V současnosti ale víme, že tento předpoklad určitě neplatí stoprocentně).
Hlavními výhodami analýzy isozymů ve srovnání s DNA metodami je nižší cena, kodominantní charakter, takže je možné přímo rozlišit heterozygoty (z DNA metod má tuto vlastnost pouze analýza mikrosatelitů) a značná univerzálnost metody. Určitou výhodou je i menší náročnost na některé aspekty práce v laboratoři (menší riziko kontaminací, netřeba pracovat sterilně, pracuje se s většími objemy roztoků). Značnou nevýhodou je naopak
Isozymy
28
nutnost izolovat isozymy z čerstvého živého materiálu. Určitou nevýhodou je i vyšší množství materiálu pro izolaci (problém u malých organismů, např. u mechorostů) a značná citlivost enzymů na teplotu a z toho vyplývající nároky na skladování a dostatečné chlazení při vlastní laboratorní práci. Pro interpretaci výsledků je také potřeba brát v úvahu, že ne všechny změny v kódující sekvenci DNA se projeví ve struktuře enzymu a ne všechny formy enzymu jsme schopni oddělit elektroforézou, takže analýza isozymů dává nižší odhady variability než je variabilita na úrovni sekvence DNA (odhady se pohybují mezi 20–60%).
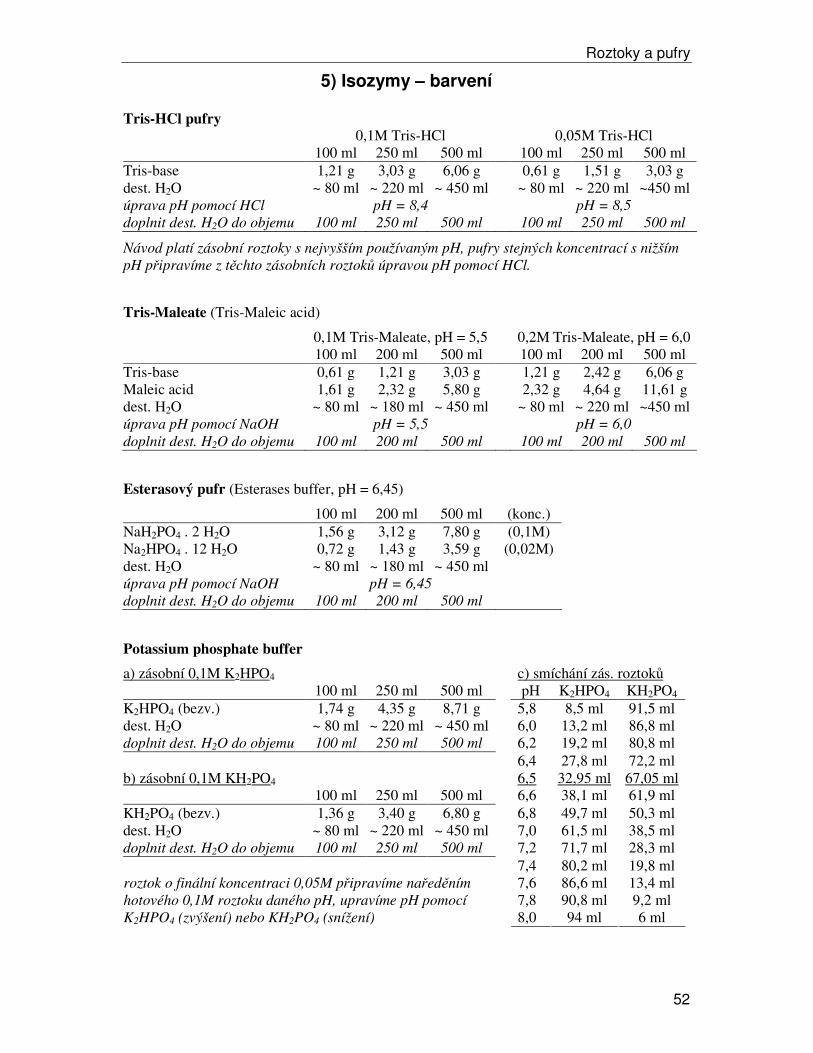
Histochemické barvení
Nedetekujeme přímo molekulu enzymu jako takovou (tj. nebarvíme přímo proteiny), ale detekujeme projevy reakce enzymem katalyzované. Barvicí roztoky mají tři hlavní součásti: (a) pufr udržující optimální podmínky pro daný enzym (určitá hodnota pH, u některých enzymů se přidává MgCl2, apod.), (b) substrát, který je specifický pro studovaný enzymový systém a je jeho činností metabolizován a (c) látky, které vytvářejí barevnou reakci. Jsou tři hlavními způsoby detekce: a) Přímá detekce. Dochází ke změně bezbarvého rozpustného substrátu na barevnou sraženinu, které se v gelu hromadí v místě aktivity enzymu a vytváří barevnou zónu. b) Nepřímá detekce. Dochází ke změně barevného substrátu na bezbarvý produkt – gel je zbarvený, pouze v místě aktivity enzymu vznikají bezbarvé pruhy. c) Spřažené reakce (využívané u naprosté většiny enzymových systémů). Produkt reakce není barevný, ale reakci je možné zviditelnit pomocí dalších souběžně probíhajících reakcí. Typickým příkladem je tzv. tetrazoliový systém používaný pro detekci většiny dehydrogenáz – ty oxidují substrát a za účasti několika přenašečů se redukuje látka světle žlutá rozpustná tetrazoliová sůl MTT (thiazolyl blue tetrazolium bromide; thiazolylová modř) na modrofialový nerozpustný formazan, viz schéma. U některých systémů není ale možná ani podobná detekce pomocí spontánně probíhajících spřažených reakcí. V takovém případě se do barvícího roztoku přidává další enzym (někdy i více), který metabolizuje produkt námi zkoumaného enzymu, přičemž reakci tohoto druhého enzymu už obarvit schopni jsme – a protože substrát tohoto druhého enzymu v kaskádě je pouze tam, kde jej vyrábí první enzym, barevná reakce označuje polohu prvního enzymu v kaskádě, viz schéma. Histochemické barvení isozymů je poměrně citlivé na přesné dodržení reakčních podmínek (teplota, pH a koncentrace roztoků, někdy je nutné postupně doplňovat substrát). Používaná barviva většinou citlivá na světlo, takže příprava barvících roztoků a vlastní barvení musí probíhat ve tmě. Velmi důležitá je i správná délka barvení, kterou je třeba pro každý organismus a enzymový systém empiricky vyzkoušet – nesmí být ani příliš krátká (proužky slabé) ani příliš dlouhá (tzv. „přebarvení“, proužky jsou pak široké a neostré, může se zvýšit zabarvení pozadí gelu a proužky jsou pak málo zřetelné).
Výsledkem analýzy isozymů je soustava proužků na gelu. Nejjednodušším vyhodnocením je pouhé porovnání pattern jednotlivých vzorků – shodují-li se genotypy (dané pozicí všech proužků) nebo ne. Takové jednoduché vyhodnocení je možné v některých případech použít, např. pro identifikaci klonů, ale obvykle nestačí. Pokud je to možné, snažíme se o tzv. alelické hodnocení – pokoušíme se najít, kolika alelami je daný lokus v studovaném souboru jedinců zastoupen, kteří jedinci jsou homozygotní a kteří heterozygotní, apod. Pro alelické hodnocení je důležité znát kvartérní strukturu studovaného enzymu a ploidní úroveň zkoumaného organismu – haploid má vždy v jednom lokusu jedinou alelu (u rostlin se s tím lze setkat u mechorostů), diploid má u heterozygotů v jednom lokusu dvě alely, tetraploid dvě, tři nebo i čtyři, atd. Druhou podstatnou informací je kvartérní struktura studovaného enzymu. Nejjednodušší situace je u monomerních enzymů, tj. enzymů, jejichž molekula je tvořena jediným proteinovým řetězcem, jedinou podjednotkou. V tomto případě odpovídá jeden proužek jedné alele – u homozygota bude vždy jeden, u heterozygota dva (u diploidů). U dimerních enzymů složených ze dvou podjednotek tvoří u diploidů pattern buď jeden proužek u homozygotů nebo tři proužky u heterozygotů (u diploidů). Tři proužky u heterozygota vznikají tak, že krajní proužky odpovídají molekulám složeným ze dvou stejných podjednotek (jednoho nebo druhého typu) a prostřední proužek odpovídá molekulám složeným ze dvou různých podjednotek. Intenzita proužků je v ideálním případě 1:2:1 – nejčastěji se kombinují různé podjednotky, lze odvodit z jednoduché kombinatoriky. Podobně lze odvodit i stav pro složitější systémy (tetramery, hexamery, …). U polyploidů počet proužků roste. I v nejjednodušším případě monomerního enzymu může mít heterozygot u tetraploida až čtyři proužky, v případě dimerů je to u tetraploida až deset proužků (při čtyřech různých alelách), u složitějších systémů ještě více. Takto složitá pattern už není obvykle možné alelicky přečíst („rozšifrovat“) a většinou se ani nedaří je obarvit – jsou zde velké rozdíly v intenzitě jednotlivých proužků, takže při dostatečném obarvení nejintenzivnějších jsou ty nejslabší nezřetelné a naopak dostatečné obarvení slabých proužků vede k přebarvení těch intenzivních.
Příklady ideálních isozymových patterns, které se mohou vyskytnout u jednoho jedince určité ploidie a kvartérní struktury enzymu. U tetraploidů je zobrazen jen výběr ilustrující možné počty a intenzity proužků, ale ne všechny genotypy (analogicky budou vypadat např. bbbb, bbbc, aadd, atd.), vzájemné polohy jednotlivých alel mohou být samozřejmě jiné a u složitějších pattern se některé proužky mohou překrývat.
Isozymy
30
Interpretaci isozymových pattern mohou komplikovat i další faktory: (a) Různá intenzita u různých jedinců. Rozdíly v celkové intenzitě barvení mezi jedinci většinou nelze brát v úvahu. Nemusí nesouviset s genetickými rozdíly, ale se způsobem zpracování vzorků, s rozdílným stavem, v jakém byly jednotlivé rostliny v době odběru nebo přesném množství izolovaného materiálu. Příliš rozdílná intenzita je problém při barvení, část jedinců bude nedobarvených nebo přebarvených a jejich pattern nečitelná, takové vzorky je obvykle nutné analyzovat znovu (a např. nanést na gel úměrně větší nebo menší množství extraktu). Někdy ale může mít intenzita (resp. až absence) barvení určitý význam, např. u jednoho taxonu se určitý lokus barví a u druhého ne. (b) Sekundární proužky. U některých systémů se objevuje více proužků i u homozygotů. Tyto sekundární proužky mohou být nezřetelné nebo neostré („duchy“) nebo méně intenzívní, ale někdy jsou intenzitou nebo ostrostí srovnatelné se skutečnými „hlavními“ proužky. Mohou vznikat např. degradací enzymu, která vede k odlišné pohyblivosti při elektroforéze (přičemž ale i degradovaná molekula má nějakou enzymatickou aktivitu) nebo může jít o důsledek posttranslačních úprav. (c) Nulové alely. Někdy se vyskytují formy enzymu, které nemají žádnou nebo téměř žádnou katalytickou aktivitu a v gelu se nebarví (jakoby tam nebyly). Obvykle nemohou být v homozygotním stavu (takový organismus by měl narušený základní metabolismus), ale mohou existovat u heterozygotů. U monomerních enzymů je není možné přímo odhalit (projevit se mohou ale mohou např. u analýzy kříženců, pokud pattern u potomstva „nesouhlasí“ s pattern rodičů). U dimerů se projevují tak, že (u diploidů) je pattern tvořeno dvěma proužky místo tří, přičemž heterodimerní („střední“) proužek je slabší. Analogicky to platí u složitějších systémů a nebo polyploidů. (d) Intergenické (interlokusové) proužky. U enzymů složených z více podjednotek se mohou někdy skombinovat podjednotky z různých lokusů a vzniklá molekula je přesto funkční (i když je aktivita obvykle slabá). Může jít například o dvě kopie genu pro týž enzym, jednu jadernou a jednu z organel – v živé buňce se proteiny podle nich vytvořené asi nikdy nesetkají, ale v homogenátu pro isozymovou analýzu to možné je. V tomto případě nacházíme heterodimerní proužky i mezi lokusy.
Extrakce vzorků
a) odběr vzorků v terénu Isozymy izolujeme vždy z čerstvého živého materiálu. Ideální materiál je u každého druhu třeba vyzkoušet, protože aktivita jednotlivých enzymů se mezi jednotlivými orgány může lišit a může se dosti měnit během ontogenetického vývoje. Nejčastěji se používají mladé listy. Pro izolaci potřebujeme v typickém případě okolo 0,75 g tkáně, což obvykle odpovídá 1 až několika málo cm2 listu. Je třeba dávat pozor, aby odebraná tkáň byla zdravá, bez parazitů (houby, hmyz), a v dobrém fyziologickém stavu (nezvadnutá, nepřemrzlá, apod.). Ihned po odběru ukládáme vzorky do vlhkého a chladného prostředí, dbáme na to, aby nezvadly, nevyschly a nepřehřívaly se. Různé druhy jsou na stav tkáně při odběru různě citlivé, u těch nejcitlivějších je třeba sbírat v optimálních podmínkách (např. ráno, dokud je chladno a vlhko, může vadit i mírné zavadnutí přes poledne).
Vzorky co nejrychleji dopravíme do laboratoře k dalšímu zpracování a celou dobu se
Isozymy
31
je snažíme držet v chladnu. Typický postup při odběru v terénu je následující:
• List ihned po utržení zabalíme do vlhkého ubrousku nebo toaletního papíru
• Vzorek vložíme do igelitového pytlíku („zip-lock“) a ihned popíšeme
• Vložíme do termosky s ledem (lépe větší kusy než drobná tříšť).
• Transportujeme do laboratoře. Obvykle je možné v termosce uchovat vzorky v použitelném stavu maximálně 2–3 dny. V laboratoři před zpracování uchováváme vzorky v lednici.
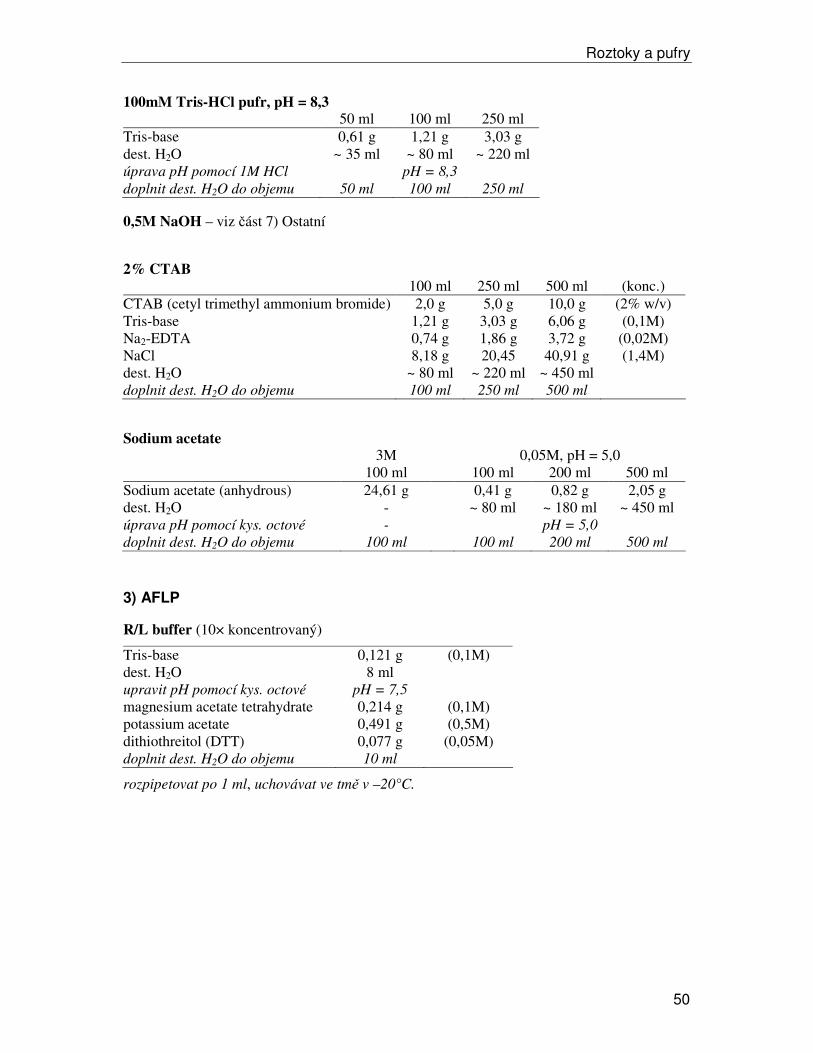
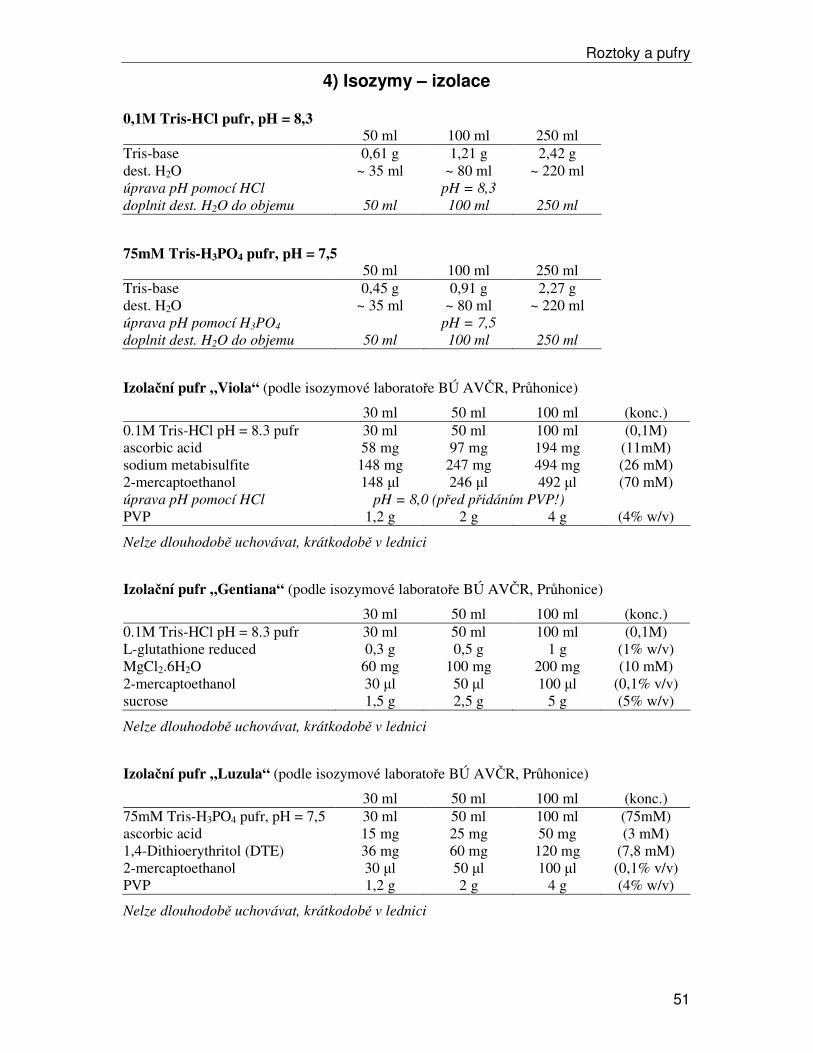
b) izolace v laboratoři Vzorky izolujeme v pufru, který stabilizuje pH a obvykle obsahuje antioxidanty a další látky, které stabilizují proteiny a umožní uchovat enzymatickou aktivitu. Izolačních pufrů existuje celá řada, je třeba vždy vyzkoušet nejvhodnější pro zkoumaný materiál (předpisy pro 3 nejčastěji používané v naší laboratoři viz Roztoky a pufry). Někdy je dobré přidat ke vzorku malé množství látky Dowex-Cl, optimální množství je třeba vyzkoušet (obvykle snižuje intenzitu pozadí na gelech, ale může snižovat i aktivitu enzymů). Celá procedura vyžaduje důkladné chlazení. Pracujeme na ledu. Před začátkem izolace dáme vychladit centrifugu na 4°C a vychladíme třecí misky a tloučky (delší dobu v lednici, při opakovaném použití během izolace alespoň krátce vychladíme v mrazáku při –20°C).
Postup izolace:
• Do vychlazené třecí misky v ledové tříšti vložíme 0,6–0,8 g rostlinné tkáně
• Přidáme 750 µl vychlazeného izolačního pufru (uchováváme v kádince v ledové tříšti) a případně Dowex-Cl.
• Tloučkem homogenizujeme rostlinný materiál. U tuhých listů může pomoct nastříhání na malé kousky nebo přidání trochy sterilního písku.
• Homogenát přepipetujeme do 1,5 ml eppendorfky. Obvykle je nutné ustřihnout asi 3 mm špičky, abychom získali širší otvor, úzký by se ucpával.
• Centrifugujeme v centrifuze vychlazené na 4°C 10 minut při 13800 rpm.
• Supernatant rozpipetujeme do 0,5 ml eppendorfek po 160 µl (dávka pro 5 vzorků + 10 µl rezerva), obvykle vychází dvě série přesně po 160 µl a třetí série s proměnlivým objemem.
• Vzorky buď hned dále zpracováváme nebo zamrazíme v –80°C (v tomto stavu lze uchovávat rok i déle; pro krátkodobé skladování lze použít i –20°C).
Elektroforéza a barvení
Pro isozymovou analýzu se používají buď horizontální škrobové nebo vertikální polyakrylamidové gely. V naší laboratoři používáme kvůli lepšímu rozlišování i delšímu uchovávání polyakrylamidové gely.
Pozor! Akrylamid je před zpolymerováním velmi jedovatý (neurotoxin), a to jak při styku s kůží, tak při vdechnutí (např. při vážení). Při manipulaci s ním a s jakýmikoliv předměty, které s ním přišly do styku (skla, stojánek, apod.) dbáme zvýšené opatrnosti a pracujeme zásadně v rukavicích a plášti, při vážení navíc s respirační rouškou. Veškeré kontaminované předměty co nejdříve oplachujeme větším množstvím vody. Odpadní roztoky obsahující akrylamid (např. nezpolymerovaný zbytek roztoku pro přípravu gelu) musí být skladovány ve zvláštní láhvi a zlikvidovány jako nebezpečný odpad, nikdy je proto
Isozymy
32
nevyléváme do běžného odpadu. Polyakrylamid je již netoxický, ale protože polymerace nikdy neproběhne dokonale a na gelech i aparatuře mohou být zbytky nezpolymerovaného akrylamidu, pracujeme i s hotovými gely zásadně v plášti a v rukavicích.
Pozor! Enzymy jsou velmi citlivé na teplo, proto je nutné elektroforézu důkladně chladit. Vzhledem k provizornímu chlazení v naší laboratoři (i vzhledem k délce elektroforézy) je dobré připravit gely a aparaturu (kroky a–c) odpoledne a nechat přes noc vychladit v lednici a elektroforézu a barvení provádět další den.
a) Sestavení skel (do tzv. sendvičů) Pozor! Skla pro elektroforézu jsou velmi křehká a zároveň drahá, proto s nimi zacházíme velmi opatrně. Je nutné, aby nebyly poškozené hrany a rohy, aparatura by pak netěsnila.
• Aparatura v laboratoři (Hoefer 600SE) umožňuje používat najednou 1, 2 nebo 4 gely.
• Do nalévacího stojánku umístíme těsnící vložky (silikonovou „gumovou“ stranou nahoru), stojánek postavíme na vodorovný stůl. Stojánek umístíme do misky, která by zachytila vytékající akrylamid v případě, že by skla byla špatně sestavená a netěsnila.
• Skla zkontrolujeme, pokud jsou znečištěná, omyjeme destilovanou vodou a vysušíme nebo necháme oschnout. Před sestavením vždy otřeme lihem (stačí denaturovaný), necháme uschnout. Čistá skla bereme pouze za hrany a nepokládáme je přímo na stůl (lze na podložku z buničiny nebo filtrační papír).
• Spacery a hřebeny omyjeme lihem, necháme uschnout.
• Na kratší strany vnějšího skla (bez výřezu)položíme pár spacerů, na ně položíme vnitřní sklo (divider plate, tenčí sklo s výřezem), na něj opět pár spacerů a na ně druhé vnější sklo. (Tento postup platí pro přípravu čtyř gelů najednou. Při dvou gelech se vynechává krok s vnitřním sklem a druhou sadou spacerů; při použití jediného gelu se druhý sendvič nahrazuje speciální skleněnou deskou.)
• Z boku nasadíme na připravený sendvič upínací nástavce s povolenými šrouby a krajní šrouby lehce dotáhneme.
• Zvedneme sendvič do vertikální polohy, postavíme spodní stranou na stůl, povolíme šrouby a srovnáme skla a spacery. Spacery musí být dotlačené úplně do boku na kraj skel a nesmí přesahovat nahoru nad skla. Lehce přitáhneme krajní šrouby, skla otočíme o 180° a postup opakujeme s horní hranou. Opakujeme tak dlouho, než jsou spacery zcela na kraji a spodní a horní hrany skel a spacerů zcela v rovině. Pozor! Důkladně zkontrolujeme, jakákoliv nerovnost by mohla vést k netěsnosti aparatury a vytékání akrylamidu nebo elektrodového pufru.
• Přiměřeně (ne velkou silou) dotáhneme všechny boční šrouby; postupujeme po obou hranách zároveň, nikdy křížem (skla by mohla prasknout).
• Spodní hranu skel namažeme vazelínou (při rutinním zvládnutí postupu lze vynechat) a skla postavíme do stojánku.
• Z boku do otvorů na stojánku vložíme utahovací kolíky, delším koncem dolů.
• Plynulým pohybem utáhneme kolíky (oba najednou a stejným směrem) plynulým pohybem směrem nahoru o 180°, skla se mírně „zaboří“ do podložky.
Isozymy
33
b) Příprava gelů
• Pro analýzu isozymů se používáme diskontinuální gely složené ze spodního 8,16% separačního gelu a 4% zaostřovacího (koncentračního gelu).
• Připravíme si 10% roztok APS (ammonium persulfate), který nelze dlouhodobě skladovat a je třeba připravit vždy čerstvý. Do 1,5 ml eppendorfky navážíme na analytických vahách množství podle počtu gelů a rozpustíme v destilované vodě.
1 gel 2 gely 4 gely APS ................................................ 0,022 g .............. 0,033 g ........... 0,067 g destilovaná voda .............................. 200 µl ................. 300 µl .............. 600 µl
• Na vnější sklo uděláme fixem značku 3,5 cm od horního okraje.
• Do vysoké 250 ml kádinky připravíme následující směs podle množství gelů:
Pufr pro separační gel ..................... 6,25 ml .............. 12,5 ml ........... 22,5 ml Zásobní roztok Akrylamid + BIS ..... 4,4 ml ............... 8,75 ml .......... 15,75 ml destilovaná voda ............................. 10,6 ml ............. 21,25 ml ......... 38,25 ml
• Opatrně mícháme kroužením (netřepat a nedělat bubliny, kyslík brzdí polymeraci) tak dlouho, dokud roztok nebude homogenní (nebudou v něm viditelná „vlákna“ akrylamidu).
• Přidáme poslední dvě látky, které spouští a katalyzují polymeraci:
• Nabereme roztok do stříkačky, přiložíme na okraj skel a spustíme jednu větší kapku po spaceru, až dosáhne dna skel, plynulým proudem naléváme roztok až po značku. Při nalévání kontrolujeme, jestli nevznikají při hladině bubliny (pokud ano, vyženeme je na hladinu a necháme prasknout).
• Po nalití všech gelů ihned převrstvíme roztok akrylamidu destilovanou vodou. Nabereme vodu do 5 ml špičky a pomalu vypouštíme po jednom spaceru. Voda se nemísí s roztokem akryalmidu a vytváří klín na hladině, přestaneme přidávat, až špička klínu dosáhne asi 1 cm od druhého spaceru.
• Necháme gel nejméně 1 hodinu polymerovat. Stejně tak necháme polymerovat zbytek roztoku v kádince, nevyléváme ho do odpadu!
• Po zpolymerování separačního odsajeme injekční stříkačkou vodu nad gelem. Protože obsahuje i zbytky nezpolymerovaného akrylamidu, zacházíme s ní jako s nebezpečným odpadem! Stejně zacházíme s nezpolymerovaným zbytkem v kádince!
• Do vysoké 100 ml kádinky připravíme následující směs pro koncentrační gel: 1 gel 2 gely 4 gely
Pufr pro koncentrační gel ................. 4,5 ml ................. 9 ml ............... 18 ml Zásobní roztok akrylamid + BIS ...... 0,5 ml ................. 1 ml ................ 2 ml
• Opatrně mícháme kroužením (netřepat a nedělat bubliny, kyslík brzdí polymeraci) tak dlouho, dokud roztok nebude homogenní (nebudou v něm viditelná „vlákna“ akrylamidu).
• Přidáme poslední dvě látky, které katalyzují polymeraci: 10% APS .......................................... 30 µl .................. 60 µl .............. 120 µl 10% TEMED .................................... 30 µl .................. 60 µl .............. 120 µl
Isozymy
34
• Opatrně promícháme a ihned naléváme gel
• Nabereme roztok do stříkačky a naléváme do úrovně výřezů ve vnitřním skle (resp. asi 5 mm pod okraj skel při nalévání jediného gelu v sendviči)
• Mezi skla zastrčíme hřebeny. Při použití čtyř gelů je dobré srovnat hřebeny tak, aby jamky v rámci jednoho sendviče byly přesně za sebou (usnadní to nanášení vzorků).
• Zkontrolujeme, jestli nezůstaly bubliny pod zuby hřebenů, pokud ano, vyženeme je pohybem nebo nadzvednutím hřebene.
• Necháme gel nejméně 1 hodinu polymerovat. Stejně tak necháme polymerovat zbytek roztoku v kádince, nevyléváme ho do odpadu!
• Během polymerace si připravíme elektrodový pufr (viz protokoly „Roztoky a pufry“).
• Po zpolymerování gelu opatrně vyjmeme hřebínky (pozor na poškození jamek!). Případné zbytky roztoku co nejrychleji odsajeme injekční stříkačkou. Roztok obsahuje zbytky nezpolymerovaného akrylamidu, zacházíme s ním jako s nebezpečným odpadem!
• Jamky ihned naplníme elektrodovým pufrem (pipetou, vypouštíme prudce, jamky se proudem pufru lépe vypláchnou).
• Znovu vysajeme pufr z jamek (nyní již s ním zacházíme jako s běžným odpadem) a jamky znovu naplníme elektrodovým pufrem.
• Horní hranu gelů překryjeme alobalem, aby nevysychaly, a dáme chladit do lednice.
c) Příprava aparatury
• Spodní vanu pro elektroforézu naplníme ředěným elektrodovým tris-glycinovým pufrem (2,4 l neředěného pufru + 1,8 l destilované vody). Do vany vložíme magnetické míchadlo.
• Vanu elektroforézy umístíme do větší plastové vany a vložíme do lednice pro elektroforézu. Do vany zasuneme skleněný výměník tepla. Necháme vychladit přes noc.
• Do výřezů na spodní straně horní vany elektroforézy vložíme těsnící vložky. Výřezy nejprve postříkáme malým množstvím destilované vody, kapky rozetřeme do plochy a na ně těsnění „přilepíme“. Umístíme do lednice a necháme vychladit přes noc.
d) Nanášení vzorků a elektroforéza
• Zapneme chlazení (spínač na boku pro chlazení + zapojení pumpičky prostým zapojením druhého kabelu do elektrické sítě) a zapneme magnetické míchadlo.
• Aparaturu elektroforézy v lednici obsypeme ledem.
• Dáme rozmrazit vzorky (trvá asi 15–20 minut), po rozmrznutí uchováváme na ledu!
• Těsně před nanášením vzorků vyndáme z lednice stojánek s gely, doplníme do jamek vychlazený elektrodový pufr až po okraj.
• Pipetou nanášíme 30 µl vzorku (v případě potřeby je množství možné upravit). Pozor! Pracujeme co nejrychleji, aby se vzorky citlivé na teplo co nejméně zahřívaly.
• Po nanesení vzorků povolíme šrouby držící gely ve stojánku (plynulým pohybem k sobě oba šrouby najednou o 180°; delší konec bude nakonec mířit dolů) a vyjmeme je z otvorů.
• Na horní hranu skel nasadíme horní vanu elektroforézy, do otvorů vložíme šrouby delším koncem nahoru a přitáhneme plynulým pohybem o 180° k sobě (delší konce dolů).
• Horní vanu s gely vložíme do spodní vany tak, aby výměník tepla zůstal mezi gely.
• Nalijeme elektrodový pufr (neředěný tris-glycinový pufr) do horní vany. Pufr naléváme nejprve do středu vany a pomalu zaplavujeme štěrbiny spojující vanu s gely, nikdy
Isozymy
35
nenaléváme pufr přímo do štěrbin, aby se nevyplavily vzorky z jamek. Pufr naléváme do výšky asi 0,5 – 1 cm (ne více!) nad drátek (elektrodu) vprostřed vany.
• Vanu přikryjeme víkem, otvory ve víku musí dosednout přesně na konce elektrod.
• Připojíme elektroforézu na zdroj elektrického proudu, dbáme na správnou polaritu.
• Zapneme elektroforézu na konstantní proud 80 mA, po ustálení (cca po 1 min.) zaznamenáme do protokolu hodnotu proudu, napětí a výkonu.
• Pravidelně kontrolujeme průběh elektroforézy. Ve chvíli, kdy zbarvené čelo elektroforézy dosáhne asi 0,5 cm od spodního okraje (asi 4:30–5 hod.), vypneme zdroj elektřiny a odpojíme elektroforézu od zdroje.
• Do protokolu zaznamenáme celkovou dobu elektroforézy a koncové hodnoty proudu, napětí a výkonu.
e) Příprava barvení V průběhu elektroforézy připravíme barvicí roztoky. Pozor! Barvící roztoky jsou citlivé na světlo. Všechny látky vystavujeme světlu co nejméně, ihned po vážení je uklízíme zpět do skříně nebo lednice, navážená množství uchováváme v kádince obalené alobalem, ihned po vážení je odneseme do temné komory.
• V průběhu elektroforézy připravíme barvicí roztoky.
• Na analytických vahách navážíme látky do barvícího roztoku podle receptů pro jednotlivé enzymové systémy. Pracujeme vždy v rukavicích, některé z používaných látek jsou jedovaté. Navážené látky přesypeme do 100 ml kádinek obalených alobalem.
• Pokud je součástí postupu přidání enzymu do barvicího roztoku, nepřidáváme jej nikdy během vážení, ale až těsně před aplikací roztoku na gel.
• Navážené směsi skladujeme v temné komoře.
• Odměříme si příslušné objemy pufrů. Používáme připravené zásobní roztoky dané koncentrace, kde je potřeba, upravíme pH.
• Asi 10 minut před koncem elektroforézy rozpustíme navážená barviva v příslušných pufrech (někdy rozpouštění několik minut trvá, je třeba barviva důkladně rozmíchat).
• Těsně před skončením elektroforézy připravíme barvicí misky, do kterých dáme vychlazenou destilovanou vodu nebo vychlazený pufr pro opláchnutí gelů (podle enzymového systému).
f) Vyjmutí gelů
• Vytáhneme gely z aparatury, pufr z horní vany vylijeme do spodní vany nebo do odpadu.
• Gely umístíme do stojánku, povolíme šrouby a odstraníme horní vanu.
• Sendvič položíme na stůl, povolíme šrouby na upínacích nástavcích a ty odděláme, skla opatrně položíme na filtrační papír na stůl.
• Plastovou špachtlí (tenčím koncem) nebo nožem zatlačíme na spodní stranu spacerů a vytáhneme je. Snažíme se nepoškodit gel se vzorky.
• Širší konec plastové špachtle přiložíme asi do poloviny boční strany skel do mezery po spaceru (případně přiložíme nůž po celé délce skel), druhou stranu skla přidržujeme prsty a opatrně zapáčíme, dokud se skla neodlepí od sebe. Pokud gel zůstane přilepený na horním skle, rychle sklo otočíme gelem nahoru a položíme na stůl.
• Odřízneme koncentrační gel a šikmým ukrojením pravého spodního rohu označíme pozici posledního vzorku (pozor na opačnou orientaci gelu, pokud zůstal na horním skle!).
Isozymy
36
• Navlhčíme si rukavice, nadzvedneme jeden okraj gelu, uchopíme do prstů a plynulým pohybem zvedneme ze skla a přeneseme do připravené barvicí misky.
g) Barvení
• Gely v barvicích miskách necháme chvíli oplachovat.
• Slijeme destilovanou vodu nebo oplachovací pufr, gely v miskách přeneseme do temné komory a (za minimálního osvětlení) nalijeme do misek barvicí roztok. Pokud je součástí postupu přidání enzymu, přidáme jej těsně před nalitím roztoku na gel.
• Promícháme a vložíme do termostatu.
• Průběžně promícháváme barvící roztok a kontrolujeme (za minimálního osvětlení!) stav barvení, abychom gely nepřebarvili.
• Po skončení barvení slijeme barvicí roztok do k tomu určené nádoby (jde o nebezpečný odpad, nelze vylévat do běžného odpadu!). Gel opláchneme v destilované vodě.
• Pokud je to potřeba (jen některé enzymové systémy), fixujeme gel ve fixačním roztoku. Po skončení fixace vylijeme fixační roztok do nádoby na nebezpečný odpad, nevyléváme jej nikdy do běžného odpadu!
• Gel oplachujeme v destilované vodě, kterou průběžně měníme, dokud se z gelu nevymyjí zbytky barvícího (příp. fixačního) roztoku.
Vysušení gelu
Akryalmidové gely lze vysušit mezi dvěma vrstvami celofánu a ± trvale uchovávat ve vysušeném stavu:
• Do fotografické misky nalijeme 1–2 cm destilované vody (podle použitých rámečků).
• Na dno misky vložíme rámeček.
• Vezmeme list celofánu a oplachujeme pod mírně tekoucí vodou, dokud nezměkne.
• Rozprostřeme list celofánu na rámeček tak, aby pod ním ani nad ním nezbyly žádné bubliny (případné bubliny vyženeme na hladinu a necháme prasknout).
• Na celofán položíme gel.
• Opláchneme druhý list celofánu a opatrně přiložíme na spodní celofán a gel. Mezi oběma vrstvami celofánu ani okolo gelu nesmí zbýt žádné bubliny (potrhaly by vysýchající gel).
• Přiložíme horní rámeček.
• Srovnáme gel a obě vrstvy celofánu tak, aby vyčnívaly z rámečků na všechny strany stejně daleko a posuneme gel do středu rámečku.
• Ohneme obě vrstvy celofánu přes jednu delší stranu rámečku, vyženeme bubliny (nesmí se dostat mezi rámečky ke gelu) a přichytíme celofán k rámečkům kolíčky na prádlo. Napneme celofán a pokračujeme s dalšími dvěma stranami.
• Narovnáme gel do středu rámečků, podepřeme jej prsty (aby se nesesunul) a zvedneme rámečky tak, aby poslední volná strana zůstala dole. Vypustíme přebytečnou vodu. Vrátíme rámečky zpět do misky a přichytíme kolíčky i poslední stranu.
• K rámečkům připneme popisek (enzymový systém, datum zpracování) a necháme gel vyschnout. Při použití tenkých rámečků je ještě musíme po asi 30 min. až 1 hodině přepnout na pevnou podložku, aby se vysýchající celofán nezkroutil.
Isozymy
37
• Po cca 2 dnech můžeme vysušený gel vyjmout z rámečků. Ostřihneme celofán tak, aby zbyl okolo gelu asi 1 cm široký pruh, na který napíšeme čísla vzorků a studovaný druh, datum a enzymový systém.
• Gel necháme několik dnů lisovat. Gely dále uchováváme v suchu a zbytečně nevystavujeme světlu (pokud s gelem zrovna nepracujeme, uchováváme jej v neprůhledných deskách ve skříni nebo šuplíku, ve tmě).
Jednotlivé enzymové systémy
V této části jsou uvedeny detaily o jednotlivých enzymových systémech a návody na barvení. Uvádíme vždy (pokud jsou informace v literatuře dostupné): - zkratku enzymu a název (v angličtině, české názvy jsou stejné, jen s českými koncovkami) - oddělené pomlčkami: E.C. číslo, kvarterní strukturu, lokalizaci v buňce (c – cytosol, m –
mitochondrie, p – plastidy) a počet lokusů - praktické poznámky, upozornění na možné komplikace, apod. - složení barvicích roztoků (názvy chemikálií z praktických důvodů anglicky); návody na
pufry viz Roztoky a pufry - informace k barvení (teploty, odchylky od standardního postupu, apod.)
ACP – acid phosphatase
E.C. 3.1.3.2 – monomer, dimer – c, m, p – 2 až 4
0,05M sodium acetate buffer, pH = 5.0 30 ml (+ asi 50 ml na oplachování) 1-naphthylphosphate 50 mg Fast Black K salt 5 mg
Po skončení elektroforézy neoplachujeme kvůli pH v destilované vodě, ale v předem vychlazeném pufru (je proto třeba proto připravit asi 100 ml pufru). Barvení při 37°C ve tmě
ADH – alcohol dehydrogenase
E.C. 1.1.1.1 – dimer – c – 1 až 3 často variabilní; dosti citlivý na teplotu; někdy se na gelu negativně barví také SOD
Přidat ke gelu roztok A, inkubovat ve tmě při 32°C, po 2-3 minutách přidat ethanol (roztok B). V případě, že se gel barví slabě, přidávat další dávky ethanolu vždy po 1 hodině; možné je i prodloužit úvodní inkubaci bez ethanolu až na 10 minut.
E.C. 2.6.1.1 – dimer – c, m, p – až 4 často variabilní; častý výskyt sekundárních proužků, které mohou komplikovat alelické vyhodnocení; poměrně citlivý na teplotu
Roztok B: 0,1M Tris–HCl buffer, pH = 8.4 20 ml pyrodoxal-5-phosphate 25 mg Fast Blue BB salt 50 mg Fast Violet B salt 50 mg
Roztok A připravovat na světle dostatečně dlouho (min. 15 minut) předem, při rozpouštění nutno zahřát na cca 50°C, poté nechat zchladnout na pokojovou teplotu. Roztoky A a B smícháme těsně před inkubací ve tmě (!) a ihned nalijeme na gel. Inkubace ve tmě při 32°C. Po inkubaci slijeme barvicí směs, krátce gel opláchneme destilovanou dobou a fixujeme fixačním roztokem. Dobu fixace je nutno vyzkoušet, u některého materiálu možno přes noc, někdy ale začínají proužky po určité době (někdy jen 15 min.) slábnout, v té chvíli je nutno fixaci ukončit. Po skončení fixace slijeme fixační roztok (obsahuje methanol – nebezpečný odpad!) a gel oplachujeme v destilované vodě, kterou průběžně měníme (dokud je z gelu zřetelně cítit kys. octová).
DIA – diaphorase
E.C. 1.6.-.- – monomer, dimer, tetramer – c, m – 1 až 4 barví se většinou poměrně rychle, pozor na přebarvení; obvykle alespoň jeden dobře čitelný monomerní lokus + nějaké další, čtení ale někdy komplikováno sekundárními pruhy
0,1M Tris–maleic acid buffer, pH = 5.5 50 ml (+ asi 50 ml na oplachování) BANA 5 mg (β-N-benzoyl-DL-arginine-α-naphthylamide.HCl)
Fast Black K salt 5 mg MgCl2.6H2O 25 mg
Po skončení elektroforézy neoplachujeme kvůli pH v destilované vodě, ale v předem vychlazeném pufru (je proto třeba proto připravit asi 100 ml pufru). Barvení při 37°C ve tmě.
Isozymy
39
EST – esterases
E.C. 3.1.1. – monomer, dimer – c – 2 až 10 poměrně nespecifické barvení, vždy více lokusů; dosti variabilní, vhodný např. pro identifikaci klonů, ale alelická interpretace většinou obtížná (prolínání lokusů, apod.); poměrně citlivý na teplotu
Esterase buffer, pH = 6.45 60 ml (+ asi 50 ml na oplachování) 1-naphthyl acetate 25 mg (předem rozpustit v 2,5 ml 50% acetonu) 2-naphthyl phosphate 25 mg (předem rozpustit v 2,5 ml 50% acetonu) Fast Blue BB Salt 50 mg
Gel po skončení elektroforézy neoplachujeme v dest. vodě, ale v esterasovém pufru (kvůli pH), je tedy potřeba připravit min. 100 ml pufru. Inkubace ve tmě při 32°C.
0,05M potassium phosphate buff.,pH=6,5 50 ml (+ asi 50 ml na oplachování) 6-bromo-2-naphthyl-β-D-glucoside 50 mg (rozpustit v 5 ml N,N’-dimethylformamidu) PVP-40 1 g Fast Blue BB salt 50 mg
Po skončení elektroforézy neoplachujeme kvůli pH v destilované vodě, ale v předem vychlazeném pufru (je proto třeba proto připravit asi 100 ml pufru). Inkubace ve tmě při 32°C 60 minut a potom v pokojové teplotě přes noc (ve tmě!)
0,1M Tris–HCl buffer, pH=7,1 50 ml glucose 45 mg MgCl2.6H2O 50 mg ATP 12,5 mg (disodium salt) NADP+ 12,5 mg NBT 10 mg PMS 1,5 mg glucose-6-phosphate dehydrogenase 40 µl (= 40 U, zásobní roztok 1 U / µl)
Dehydrogenasu přidáváme až jako poslední těsně do již hotového roztoku před aplikací na gel, promícháme. Inkubace ve tmě při 37°C. Existují i alternativní barvení (vyšší pH, náhrada některých složek).
LAP – leucine aminopeptidase (= AMP – aminopeptidase)
E.C. 3.4.11.- – monomer – c – 2 až 3 obvykle dosti variabilní; velmi citlivý na teplotu a kvalitu vzorku
Roztok A: 0,2M Tris–maleate buffer, pH = 6.0 30 ml (+ asi 50 ml na oplachování) MgCl2.6H2O 60 mg L-leucyl-β-naphthylamide hydrochloride 35 mg (rozpustit v 2,5 ml 50% acetonu, ve tmě)
Roztok B: 0,2M Tris–maleate buffer, pH = 6.0 30 ml Fast Black K salt 25 mg
Gel po skončení elektroforézy neoplachujeme v dest. vodě, ale v pufru (kvůli pH), je tedy potřeba připravit min. 100 ml pufru. Gel inkubujeme při 32°C nejprve pouze v roztoku A, po 10 min. přidáme roztok B.
Isozymy
41
MDH – malate dehydrogenase
E.C. 1.1.1.37 – dimer – c, m – 3
Roztok A: 0,1M Tris–HCl buffer, pH=7,5 20 ml malic acid 150 mg (po rozpuštění upravit pH pomocí Na2CO3 na 7,5)
E.C. 1.6.99.- – monomer, dimer, tetramer (?) – c, m, p – 1-4 (?) většinou je tento název používán i pro DIA, resp. nejsou oba enzymové systémy odlišovány (katalyzují téměř totéž, někdy se oběma barveními barví stejné enzymy, ale někdy ne)
0,1M Tris–HCl buffer, pH=8,4 30 ml menadion 15 mg (rozpustit v 2,5 ml 50% acetonu) NADH (reduced) 17,5 mg MTT 20 mg
Inkubace ve tmě při 37°C.
PRX – peroxidase
E.C. 1.11.1.7 – monomer – c – 2-13
0,05M sodium acetate buffer, pH=5,0 50 ml (+ asi 50 ml na oplachování) CaCl2 11 mg (anhydrous) 3% H2O2 250 µl 3-amino-9-ethyl carbazole 25 mg (rozpustit v 5 ml N,N’-dimethylformamidu)
Gel po skončení elektroforézy neoplachujeme v dest. vodě, ale v pufru (kvůli pH), je tedy potřeba připravit min. 100 ml pufru. Inkubace v lednici. Po vyvinutí proužků fixace ve fixačním roztoku.
E.C. 5.1.3.9 – dimer – c, p – 2 někdy se spolu s PGI barví další enzymy (které následují v kaskádě za ním, 6PGDH nebo G6PDH), ale interpretace je možná, pokud se lokusy nepřekrývají
Dehydrogenasu přidáváme až jako poslední těsně do již hotového roztoku před aplikací na gel, promícháme. Inkubace ve tmě při 32°C.
PGM – phosphoglucomutase
E.C. 2.7.5.1 – monomer – c, p – 2 někdy se spolu s PGM barví další enzymy (které následují v kaskádě za ním, 6PGDH nebo G6PDH), ale interpretace je možná, pokud se lokusy nepřekrývají
Inkubace ve tmě při 37°C po dobu 20 min a potom na světle (ideálně pod lampou) za laboratorní teploty až do objevení světlých proužků na tmavém pozadí. Nenechávat na světle zbytečně dlouho, časem (cca 20 min., třeba vyzkoušet) proužky začínají slábnout (pozadí se barví i do nich).
Obsluha přístrojů
44
Obsluha přístrojů
Měření koncentrace DNA pomocí spektrofotometru Biowave II
• Přístroj se zapíná/vypíná černým tlačítkem (hlavní spínač) vlevo na přední straně přístroje.
• K pohybu po displeji a výběru požadované (zvýrazněné) volby slouží šipkové klávesy.
• Číselná tlačítka vpravo na přístroji (klávesnice) slouží k psaní textu, vkládání hodnot; opakovaným stiskem se přepíná mezi malými a velkými písmeny; klávesa 1 slouží k vkládání mezery; klávesa „C“ = backspace.
• Červené tlačítko (Escape/Cancel) slouží k návratu do předchozího kroku; zastavení měření.
• Modré tlačítko (0A/100%T) slouží k nastavení blanku.
• Zelené tlačítko (Enter) slouží k potvrzení výběru a k provedení měření
• Pro měření koncentrace DNA zvolíme stisknutím příslušného čísla funkci „Life Science“
→ „Nucleic Acid“ → „DNA“
• DNA neměříme přímo, ale zředěnou (koncentrace by mohla být mimo měřitelnost přístroje a hlavně – nemusíme svojí DNA plýtvat); minimální množství roztoku v kyvetě je 70 µl
• Zadáme ředění vzorku („Dilution Factor“), např. při ředění 1:13 (5 µl DNA + 65 µl vody) je ředící faktor 14; zvolíme jednotky (ng/µl) a potvrdíme nastavení (ostatní parametry necháme v původním nastavení)
• Připravíme si referenční vzorek (blank). Bude jím buď voda nebo roztok, ve kterém je DNA skladována (Elution Buffer D z kitu, TE pufr, apod.) – ve druhém případě tento roztok ředíme vodou stejným poměrem jako budeme používat pro měření DNA.
• Vložíme kyvetu s referencí a stiskneme modré tlačítko pro nastavení blanku, přístroj ukáže nulovou absorbanci
• Při měření používáme stále stejnou orientaci kyvety (jedna strana kyvety je opatřena šipkou)
• Kyvetu několikrát vypláchneme destilovanou vodou ze střičky, kapky vody z kyvety vyklepeme na buničinu, příp. vysajeme zbytek vody ze dna kyvety pipetou. Kyvetu opakovaně proplachujeme mezi jednotlivými vzorky.
• Do kyvety pipetujeme zvolené množství vody a DNA, promícháme pipetováním, vložíme kyvetu se vzorkem do přístroje a stiskneme zelené tlačítko pro měření; přístroj ukáže koncentraci našeho originálního vzorku DNA (nikoliv koncentraci v kyvetě!)
• Zaznamenáme hodnoty z displeje
• Po skončení měření vyjmeme kyvetu a přístroj vypneme
• Kyvetu necháme odmočit v jarové vodě, vypláchneme destilovanou vodou a necháme vyschnout na kousku buničiny
Obsluha přístrojů
45
Zacházení s gradientovým termocyclerem TC-XP Bioerg
• přístroj se zapíná v zadní části přístroje vedle přívodní šňůry
• přístroj ovládáme pomocí tlačítek F1–F5 dole pod displejem; pohybujeme se pomocí šipek vlevo na přístroji, potvrzujeme Entrem
• po spuštění se na základní obrazovce objeví tři ikony: - ikona File – po stisknutí se zobrazí seznam uživatelů (Users), názvy souborů (File
name) datum vytvoření programu (Save date) - ikona System – systémová nastavení, např.: nastavení teploty víka, zapnutí/vypnutí
zvukového signálu po skončení programu - ikona Run
Vytvoření nového programu
• Pokud chceme měnit parametry stávajícího programu označeného kurzorem, stiskneme klávesu Edit
• Pro vytvoření nového programu stiskneme klávesu F2 (New File)
• Stiskem klávesy F1 (+Seg) zadáváme parametry jednotlivých kroků (tzv. segmentů) jako teplotu (Temp), čas (Time), rychlost nárůstu/poklesu teploty (tzv. Ramping rate – Ramp); přednastavená hodnota „#.#“ ve sloupci Ramp značí nejvyšší možnou rychlost nárůstu/poklesu teplot
• Stiskem klávesy F2 (+Cycle) nastavíme počet cyklů a průběh cyklu, např. from 02 to 04
• Pro tzv. touchdown PCR využijeme sloupec +Temp a +Time
• Sloupec Grad slouží pro vytvoření parametrů teplotního gradientu, číslice 12 znázorňuje gradient 12 stupňů mezi první a poslední pozicí bloku; teplota mezi jednotlivými pozicemi se nastaví automaticky, její nárůst od první k poslední pozici je progresivní
• Po vytvoření programu stiskneme klávesu F5 (Save), zadáme jméno uživatele a název vytvořeného programu a potvrdíme klávesou F3 (Save)
Spuštění programu
• Vložíme PCR zkumavky, stripy nebo destičku se vzorky.
• Zaklapneme víko bloku a dotáhneme (ne silou!)
• Stiskneme klávesu F1 (File), vyhledáme svůj uživatelský profil a po označení příslušného programu kurzorem stiskneme klávesu F5 (Run)
• Po spuštění programu se na displeji zobrazí jméno uživatele, název programu, celkový čas, zbývající čas, číslo a grafický průběh právě probíhajícího cyklu, počet cyklů, teplota víka apod.
Zastavení programu
• Po skončení programu stiskem klávesy Stop a opětovným potvrzením stiskem F1 (Stop).
Obsluha přístrojů
46
Zacházení s termocyclerem Biometra T3000 se třemi bloky
• Přístroj se zapíná/vypíná na přední straně vpravo dole tlačítkem „Power“
• přístroj ovládáme pomocí tlačítek dole pod displejem a na pravé straně, pohybujeme se pomocí šipek, potvrzujeme Entrem, mažeme tlačítkem Cancel
• základní obrazovka ukazuje několik ikon: - Ikona Info: zobrazení informací o blocích – právě činný (active) nebo volný (inactive) - Ikona Systém – informace o typu přístroje, nastavení signalizace konce programu,
navolení požadovaného jazyka, nastavení kontrastu na displeji apod. - Ikona Start/stop – zapnutí/vypnutí programu - Ikona Edit – vytvoření nového programu nebo změna parametrů již uloženého
programu
Vytvoření nového programu
• Stiskneme klávesu Edit.
• Vybereme adresář, do kterého chceme program uložit, stiskneme Enter.
• Vybereme podadresář a klikneme na Edit.
• Po zobrazení abecedy vložíme název programu a potvrdíme stiskem Name OK.
• Nastavíme teplotu víka (Lid Temperature) na 99°C a zapneme funkci předehřívání (Preheating ON), potvrdíme stiskem Enter.
• Nastavíme teplotní program – teplotu (Temp) vkládáme čísly, čas (Time) zadáváme buď v sekundách nebo lze minuty, příp. hodiny oddělit tečkou („3.30“ znamená 3 min 30 s, „1.5.0“ znamená 1 hod 5 min 0 s,); pro nastavení chlazení po skončení programu stiskneme „0“, objeví se „Pause“
• Pro nastavení cyklování, najedu na konec řádku základního cyklu a do sloupce „←“
vložíme číslo, které udává počáteční krok základního cyklu (k tomuto kroku, např. 2 se bude přístroj po každém dokončeném základním cyklu vracet a opakovat cyklus od tohoto kroku); do sloupce „#“ vložíme počet cyklů -1
• program uložím „Save pgm“
Spuštění programu
• Vložíme PCR zkumavky, stripy nebo destičku.
• Zaklapneme víko bloku a dotáhneme (ne silou!)
• Stiskneme Start/Stop, vybereme blok a po rozbalení adresáře program, který chceme spustit a stiskneme Start.
• Po spuštění programu se začne zahřívat víko, displej zobrazuje teplotu a čas právě probíhajícího kroku, teplotu víka; čas do konce programu se objeví po stisknutí „Info“.
• Ve chvíli, kdy víko dosáhne zadané teploty (standardně 99 °C), je automaticky přitlačeno na zkumavky, začíná vlastní reakce a na displeji je znázorňován průběh reakce.
• Po ukončení programu se zapne zvukový signál, povolíme víko a můžeme vyndat produkty PCR reakce.
• V průběhu PCR reakce je možné vytvářet či editovat další programy.
Obsluha přístrojů
47
Měření na pH-metru Hanna pH 211
Kalibrace
• Doporučuje se provádět nejméně 1× týdně, pro přesné měření 1× denně.
• Pro kalibraci použijeme vždy pufr pH = 7,01 a druhý (pH = 4,01 nebo 10,01) zvolíme podle toho, jestli plánujeme měření v kyselé nebo v zásadité oblasti.
• Zapneme přístroj tlačítkem vpravo pod displejem, ukáže se hodnota pH a teplota.
• Elektrodu i teploměr důkladně opláchneme destilovanou vodou ze střičky, opatrně osušíme buničinou.
• Vložíme elektrodu i teploměr do kalibračního pufru pH = 7,01. Otvor na spodní části kyvety (nad baničkou) musí být pod hladinou pufru!
• Necháme chvíli ustálit, spustíme kalibraci tlačítkem „CAL“.
• Přístroj začne kalibrovat, vlevo bliká „Not ready“. Počkáme, dokud se nápis nezmění na „Ready CFM“, potvrdíme stiskem tlačítka „CFM“.
• Šipkami (označeny �°C / �°C) vybereme hodnotu pH druhého kalibračního pufru (zobrazuje se na displeji úplně vpravo).
• Vyjmeme elektrodu i teploměr z pufru, opláchneme destilovanou vodou, vysušíme a vložíme do druhého kalibračního pufru.
• Bliká „Not ready“, počkáme, dokud se nápis nezmění na „Ready CFM“, dokončíme kalibraci stiskem tlačítka„CFM“.
• Pokud ihned nepokračujeme měřením, vypneme přístroj.
• Vyjmeme elektrodu i teploměr, opláchneme destilovanou vodou, osušíme buničinou a vložíme do čepičky s uchovávacím pufrem (Storage solution; pokud v něm nebude ponořená celá banička elektrody, doplnit).
Měření pH
• Zapneme přístroj.
• Opláchneme elektrodu i teploměr destilovanou vodou, opatrně osušíme buničinou.
• Vložíme elektrodu i teploměr do roztoku, otvor na spodní části kyvety (nad baničkou) musí být pod hladinou.
• Na displeji se zobrazuje teplota a hodnota pH. Počkáme, než se hodnota pH ustálí.
• Po skončení měření, pokud nepokračujeme dalším roztokem, vypneme přístroj.
• Vyjmeme elektrodu i teploměr, opláchneme destilovanou vodou, osušíme buničinou a vložíme do čepičky s uchovávacím pufrem (Storage solution; pokud v něm nebude ponořená celá banička elektrody, doplnit).
Roztoky a pufry
48
Roztoky a pufry
1) Elektroforéza a příprava gelů
TBE pufr (zásobní roztok)
10× konc. 20× konc. Tris-base 108 g 216 g Boric acid 55 g 110 g Na2-EDTA 9,3 g 18,6 g rozpustit v dest. H2O, doplnit do 1000 ml 1000 ml
Ředění na konc. 1× (elektroforéza, příprava gelů):
10× konc. 20× konc. 1 l 5l 1 l 5 l TBE 100 ml 500 ml 50 ml 250 ml dest. H2O 900 ml 4500 ml 950 ml 4750 ml
TAE pufr (10× konc. zásobní roztok)
1000 ml Tris-base 48,46 g Acetic acid (glacial) 12,01 g Na2-EDTA 3,72 g rozpustit v dest. H2O, doplnit do 1000 ml
Tris-glycine 1 l 3 l Tris-base 2,5 g 7,5 g Glycine 18 g 54 g dest. H2O ~ 900 ml ~ 2700 ml úprava pH pomocí HCl pH = 8,3
doplnit dest. H2O do objemu 1 l 3 l
Ředění do spodní vany elektroforézy: Tris-glycine buffer 2400 ml + dest. H2O 1800 ml
Pufr pro separační gel – isozymy (1,82M Tris-HCl, pH = 8,9)
50 ml 100 ml 200 ml Tris-base 11,05 g 22,1 g 44,2 g dest. H2O ~ 35 ml ~ 80 ml ~ 170 ml úprava pH pomocí HCl pH = 8,9 doplnit dest. H2O do objemu 50 ml 100 ml 200 ml
Pufr pro zaostřovací gel – isozymy (69mM Tris-H3PO4, pH = 6,9)
50 ml 100 ml 200 ml Tris-base 0,42 g 0,83 g 1,66 g dest. H2O ~ 35 ml ~ 80 ml ~ 170 ml úprava pH pomocí H3PO4 pH = 6,9 doplnit dest. H2O do objemu 50 ml 100 ml 200 ml
Roztoky a pufry
49
Zásobní roztok akrylamidu 100 ml 200 ml Acrylamide 40 g 80 g N,N′-Methylenebisacrylamide (BIS) 1 g 2 g rozpustit v dest. H2O, doplnit do: 100 ml 200 ml
Pozor! Obě látky jsou velmi jedovaté, pracujeme v plášti, rukavicích a při vážení v roušce!
Loading buffer
Bromphenol Blue 0,025 g Xylene Cyanol FF 0,025 g Glycerol 3 ml SYBR Green I 20 µl doplnit sterilní dest. H2O do objemu 10 ml
λ DNA Hind III digest 20 µl sterilní dest. H2O 180 µl Loading Buffer 40 µl
2) Izolace, ředění a srážení DNA
TE pufr (10× koncentrovaný zásobní roztok)
50 ml 100 ml 250 ml 1000 ml (konc.) Tris-base 0,61 g 1,21 g 3,03 g 12,11 g (0,1M) Na2-EDTA 0,19 g 0,37 g 0,93 g 3,72 g (0,1M) dest. H2O ~ 35 ml ~ 80 ml ~ 220 ml ~ 900 ml úprava pH pomocí HCl / NaOH pH = 8,0 pH = 8,0
doplnit dest. H2O do objemu 50 ml 100 ml 250 ml 1000 ml
T0.1E pufr (10× koncentrovaný zásobní roztok)
50 ml 100 ml 250 ml 1000 ml (konc.) Tris-base 0,61 g 1,21 g 3,03 g 12,11 g (0,1M) Na2-EDTA 0,019 g 0,037 g 0,093 g 0,37 g (0,01M) dest. H2O ~ 35 ml ~ 80 ml ~ 220 ml ~ 900 ml úprava pH pomocí 1M HCl pH = 8,0
doplnit dest. H2O do objemu 50 ml 100 ml 250 ml 1000 ml
Roztoky a pufry
50
100mM Tris-HCl pufr, pH = 8,3 50 ml 100 ml 250 ml Tris-base 0,61 g 1,21 g 3,03 g dest. H2O ~ 35 ml ~ 80 ml ~ 220 ml úprava pH pomocí 1M HCl pH = 8,3
doplnit dest. H2O do objemu 50 ml 100 ml 250 ml
0,5M NaOH – viz část 7) Ostatní
2% CTAB 100 ml 250 ml 500 ml (konc.) CTAB (cetyl trimethyl ammonium bromide) 2,0 g 5,0 g 10,0 g (2% w/v) Tris-base 1,21 g 3,03 g 6,06 g (0,1M) Na2-EDTA 0,74 g 1,86 g 3,72 g (0,02M) NaCl 8,18 g 20,45 40,91 g (1,4M) dest. H2O ~ 80 ml ~ 220 ml ~ 450 ml doplnit dest. H2O do objemu 100 ml 250 ml 500 ml
Sodium acetate 3M 0,05M, pH = 5,0 100 ml 100 ml 200 ml 500 ml Sodium acetate (anhydrous) 24,61 g 0,41 g 0,82 g 2,05 g dest. H2O - ~ 80 ml ~ 180 ml ~ 450 ml úprava pH pomocí kys. octové - pH = 5,0
doplnit dest. H2O do objemu 100 ml 100 ml 200 ml 500 ml
3) AFLP
R/L buffer (10× koncentrovaný)
Tris-base 0,121 g (0,1M) dest. H2O 8 ml upravit pH pomocí kys. octové pH = 7,5
magnesium acetate tetrahydrate 0,214 g (0,1M) potassium acetate 0,491 g (0,5M) dithiothreitol (DTT) 0,077 g (0,05M) doplnit dest. H2O do objemu 10 ml
rozpipetovat po 1 ml, uchovávat ve tmě v –20°C.
Roztoky a pufry
51
4) Isozymy – izolace
0,1M Tris-HCl pufr, pH = 8,3 50 ml 100 ml 250 ml Tris-base 0,61 g 1,21 g 2,42 g dest. H2O ~ 35 ml ~ 80 ml ~ 220 ml úprava pH pomocí HCl pH = 8,3
doplnit dest. H2O do objemu 50 ml 100 ml 250 ml
75mM Tris-H3PO4 pufr, pH = 7,5 50 ml 100 ml 250 ml Tris-base 0,45 g 0,91 g 2,27 g dest. H2O ~ 35 ml ~ 80 ml ~ 220 ml úprava pH pomocí H3PO4 pH = 7,5
doplnit dest. H2O do objemu 50 ml 100 ml 250 ml
Izolační pufr „Viola“ (podle isozymové laboratoře BÚ AVČR, Průhonice)
30 ml 50 ml 100 ml (konc.) 0.1M Tris-HCl pH = 8.3 pufr 30 ml 50 ml 100 ml (0,1M) ascorbic acid 58 mg 97 mg 194 mg (11mM) sodium metabisulfite 148 mg 247 mg 494 mg (26 mM) 2-mercaptoethanol 148 µl 246 µl 492 µl (70 mM) úprava pH pomocí HCl pH = 8,0 (před přidáním PVP!)
PVP 1,2 g 2 g 4 g (4% w/v)
Nelze dlouhodobě uchovávat, krátkodobě v lednici
Izolační pufr „Gentiana“ (podle isozymové laboratoře BÚ AVČR, Průhonice)
30 ml 50 ml 100 ml (konc.) 0.1M Tris-HCl pH = 8.3 pufr 30 ml 50 ml 100 ml (0,1M) L-glutathione reduced 0,3 g 0,5 g 1 g (1% w/v) MgCl2.6H2O 60 mg 100 mg 200 mg (10 mM) 2-mercaptoethanol 30 µl 50 µl 100 µl (0,1% v/v) sucrose 1,5 g 2,5 g 5 g (5% w/v)
Nelze dlouhodobě uchovávat, krátkodobě v lednici
Izolační pufr „Luzula“ (podle isozymové laboratoře BÚ AVČR, Průhonice)
30 ml 50 ml 100 ml (konc.) 75mM Tris-H3PO4 pufr, pH = 7,5 30 ml 50 ml 100 ml (75mM) ascorbic acid 15 mg 25 mg 50 mg (3 mM) 1,4-Dithioerythritol (DTE) 36 mg 60 mg 120 mg (7,8 mM) 2-mercaptoethanol 30 µl 50 µl 100 µl (0,1% v/v) PVP 1,2 g 2 g 4 g (4% w/v)
Nelze dlouhodobě uchovávat, krátkodobě v lednici
Roztoky a pufry
52
5) Isozymy – barvení
Tris-HCl pufry 0,1M Tris-HCl 0,05M Tris-HCl 100 ml 250 ml 500 ml 100 ml 250 ml 500 ml Tris-base 1,21 g 3,03 g 6,06 g 0,61 g 1,51 g 3,03 g dest. H2O ~ 80 ml ~ 220 ml ~ 450 ml ~ 80 ml ~ 220 ml ~450 ml úprava pH pomocí HCl pH = 8,4 pH = 8,5
doplnit dest. H2O do objemu 100 ml 250 ml 500 ml 100 ml 250 ml 500 ml
Návod platí zásobní roztoky s nejvyšším používaným pH, pufry stejných koncentrací s nižším
pH připravíme z těchto zásobních roztoků úpravou pH pomocí HCl.
Tris-Maleate (Tris-Maleic acid)
0,1M Tris-Maleate, pH = 5,5 0,2M Tris-Maleate, pH = 6,0 100 ml 200 ml 500 ml 100 ml 200 ml 500 ml Tris-base 0,61 g 1,21 g 3,03 g 1,21 g 2,42 g 6,06 g Maleic acid 1,61 g 2,32 g 5,80 g 2,32 g 4,64 g 11,61 g dest. H2O ~ 80 ml ~ 180 ml ~ 450 ml ~ 80 ml ~ 220 ml ~450 ml úprava pH pomocí NaOH pH = 5,5 pH = 6,0
doplnit dest. H2O do objemu 100 ml 200 ml 500 ml 100 ml 200 ml 500 ml
Esterasový pufr (Esterases buffer, pH = 6,45)
100 ml 200 ml 500 ml (konc.) NaH2PO4 . 2 H2O 1,56 g 3,12 g 7,80 g (0,1M) Na2HPO4 . 12 H2O 0,72 g 1,43 g 3,59 g (0,02M) dest. H2O ~ 80 ml ~ 180 ml ~ 450 ml úprava pH pomocí NaOH pH = 6,45
doplnit dest. H2O do objemu 100 ml 200 ml 500 ml
Potassium phosphate buffer
a) zásobní 0,1M K2HPO4 c) smíchání zás. roztoků 100 ml 250 ml 500 ml pH K2HPO4 KH2PO4 K2HPO4 (bezv.) 1,74 g 4,35 g 8,71 g 5,8 8,5 ml 91,5 ml dest. H2O ~ 80 ml ~ 220 ml ~ 450 ml 6,0 13,2 ml 86,8 ml doplnit dest. H2O do objemu 100 ml 250 ml 500 ml 6,2 19,2 ml 80,8 ml 6,4 27,8 ml 72,2 ml b) zásobní 0,1M KH2PO4 6,5 32,95 ml 67,05 ml 100 ml 250 ml 500 ml 6,6 38,1 ml 61,9 ml KH2PO4 (bezv.) 1,36 g 3,40 g 6,80 g 6,8 49,7 ml 50,3 ml dest. H2O ~ 80 ml ~ 220 ml ~ 450 ml 7,0 61,5 ml 38,5 ml doplnit dest. H2O do objemu 100 ml 250 ml 500 ml 7,2 71,7 ml 28,3 ml 7,4 80,2 ml 19,8 ml
7,6 86,6 ml 13,4 ml 7,8 90,8 ml 9,2 ml
roztok o finální koncentraci 0,05M připravíme naředěním
hotového 0,1M roztoku daného pH, upravíme pH pomocí
K2HPO4 (zvýšení) nebo KH2PO4 (snížení) 8,0 94 ml 6 ml
Roztoky a pufry
53
6) Isozymy – fixace
Fixační roztok (podle isozymové laboratoře BÚ AVČR, Průhonice)
500 ml 1000 ml Kys. octová 50 ml 100 ml Glycerol 50 ml 100 ml dest. H2O 150 ml 300 ml Methanol 250 ml 500 ml
7) Ostatní
1M HCl 100 ml 250 ml 500 ml dest. H2O ~ 70 ml ~ 200 ml ~ 400 ml konc. HCl (35%) 8,83 ml 22,07 ml 44,14 ml doplnit dest. H2O do objemu 100 ml 250 ml 500 ml
Roztoky NaOH 0,5M NaOH 1M NaOH 10M NaOH 100 ml 250 ml 100 ml 250 ml 100 ml 250 ml dest. H2O ~ 70 ml ~ 220 ml ~ 70 ml ~ 220 ml ~ 70 ml ~ 200 ml NaOH (bezvodý) 2 g 5 g 4 g 10 g 40 g 100 g doplnit dest. H2O do objemu 100 ml 250 ml 100 ml 250 ml 100 ml 250 ml
Při přípravě 0,5M NaOH pro izolaci DNA použít místo destilované vody Milli-Q vodu,
vysterilizovat v autoklávu.
1M H3PO4 100 ml 250 ml 500 ml dest. H2O ~ 70 ml ~ 200 ml ~ 400 ml konc. H3PO4 (85%) 6,74 ml 16,86 ml 33,71 ml doplnit dest. H2O do objemu 100 ml 250 ml 500 ml
1M kys. octová 100 ml 250 ml 500 ml dest. H2O ~ 70 ml ~ 200 ml ~ 400 ml kys. octová (čistá) 5,72 ml 14,30 ml 28,60 ml doplnit dest. H2O do objemu 100 ml 250 ml 500 ml