RCSB PDB Progr. NMR RCSB PDB NMR biomakromolekul Typy biomakromolekul a možnosti studia pomocí NMR ● proteiny a peptidy – rozmanité složení, omezení jen velikostí molekul ● nukleové kyseliny (RNA, DNA) a oligonukleotidy – omezení malou rozmanitostí chemického složení ● polysacharidy a oligosacharidy – omezení malou rozmanitostí chemického složení ● kombinace výše uvedených
Transcript
RCSB PDB
Progr. NMR
RCSB PDB
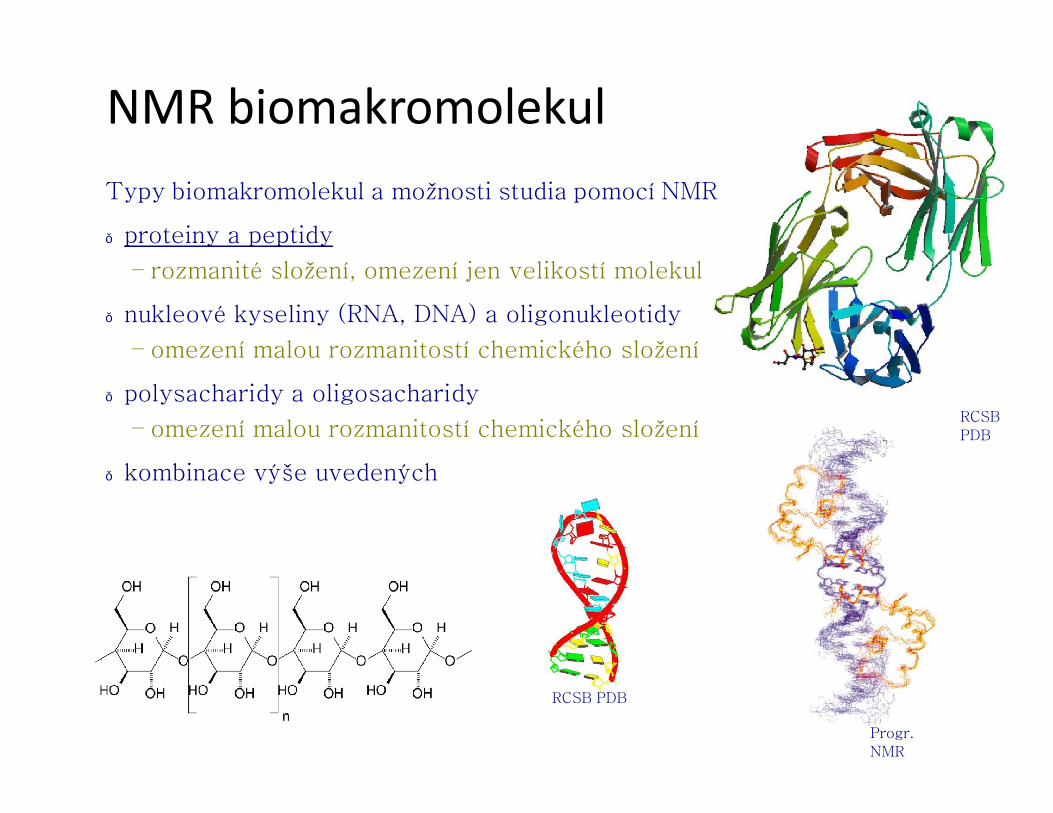
NMR biomakromolekulTypy biomakromolekul a možnosti studia pomocí NMR
● proteiny a peptidy
– rozmanité složení, omezení jen velikostí molekul
● nukleové kyseliny (RNA, DNA) a oligonukleotidy
– omezení malou rozmanitostí chemického složení
● polysacharidy a oligosacharidy
– omezení malou rozmanitostí chemického složení
● kombinace výše uvedených
NMR Proteinů
• Polymery α-aminokyselin, mnoho funkcí v organismu
• Katalytická - enzymy
• Strukturní a pohybové
• Signální
• Zásobní a transportní
• Obranné
• Pomocí NMR spektroskopie studujeme:
• Prostorové uspořádání
• Interakce s jinou molekulou
• Dynamika proteinu
Ala Asp Asn Arg Cys Glu Gln Gly His Ile
Leu Lys Met Phe Pro Ser Thr Trp Tyr Val
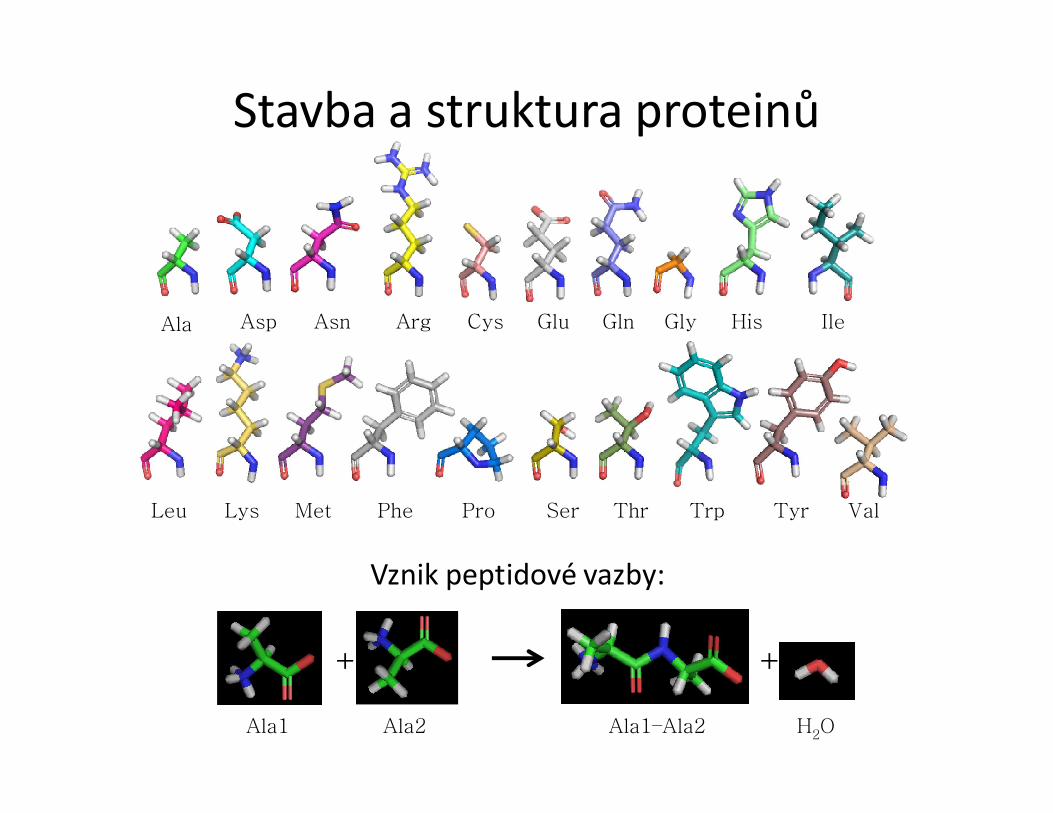
Stavba a struktura proteinů
+
Ala1 Ala2 Ala1–Ala2 H2O
+
Vznik peptidové vazby:
helikální struktura (α-helix)
extendovanástruktura (β-sheet)
náhodné klubko (random coil)
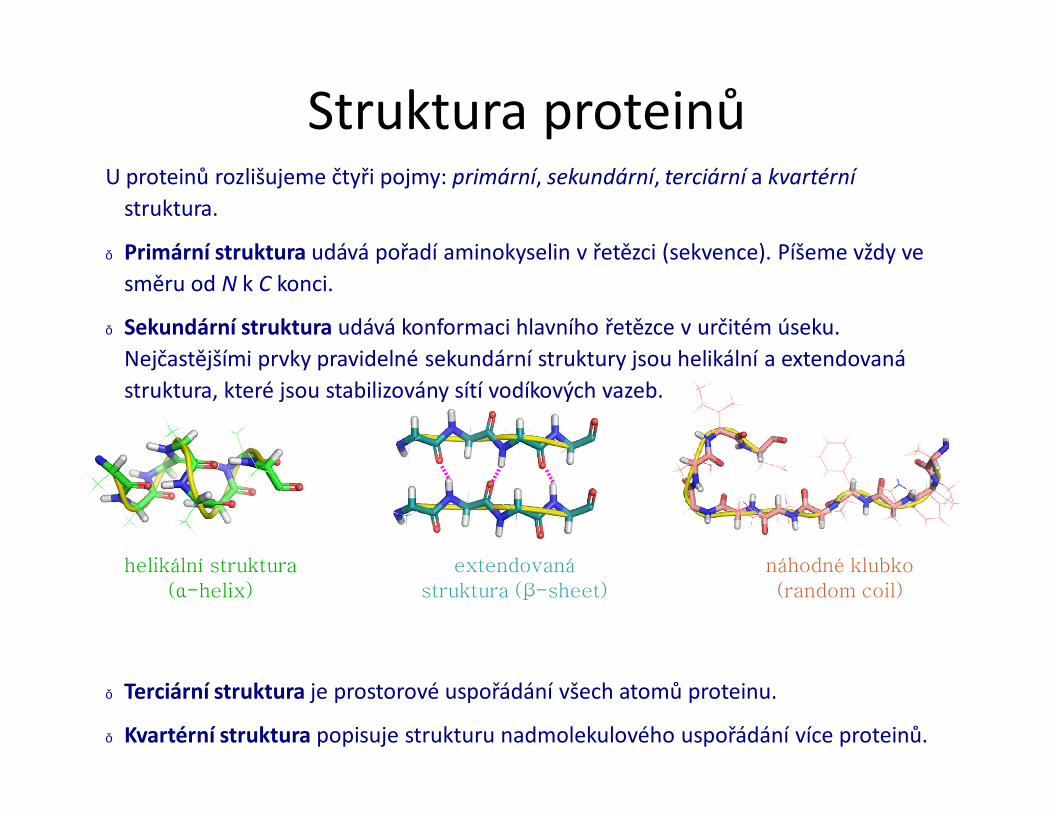
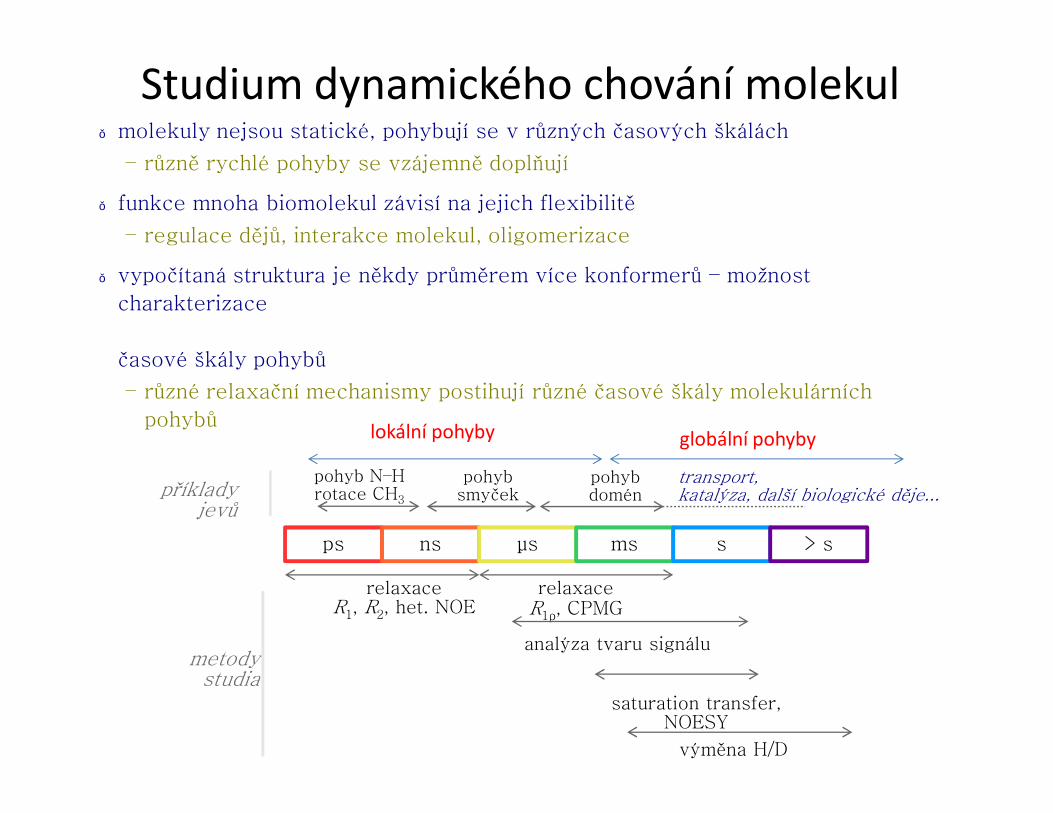
Struktura proteinůU proteinů rozlišujeme čtyři pojmy: primární, sekundární, terciární a kvartérní
struktura.
● Primární struktura udává pořadí aminokyselin v řetězci (sekvence). Píšeme vždy ve směru od N k C konci.
● Sekundární struktura udává konformaci hlavního řetězce v určitém úseku. Nejčastějšími prvky pravidelné sekundární struktury jsou helikální a extendovanástruktura, které jsou stabilizovány sítí vodíkových vazeb.
● Terciární struktura je prostorové uspořádání všech atomů proteinu.
● Kvartérní struktura popisuje strukturu nadmolekulového uspořádání více proteinů.
Zdroje Proteinů• Původní organismus• + přirozená forma včetně všech modifikací• - malé množství, drahé, nemožnost izotopového značení, etické problémy,
složitá izolace,….• Syntéza proteinu v mikroorganismech (E. coli, P. pastoris)• + levné, velký výtěžek proteinu, snadné uniformní izotopové značení• - možné problémy s modifikacemi postranních řetězců• Chemická syntéza• + velké možnosti izotopového značení, rychlé, vhodné pro toxické proteiny• - drahé, menší výtěžky, omezení maximální velikosti, problémy se
správným sbalením proteinu• In-vitro translace• + vhodné pro toxické proteiny, možnost selektivního izotopového značení• - drahé, posttranslační modifikace
Využívaná jádra v biomakromolekulách
+ vysoké přirozené zastoupení
+ vysoká citlivost (1,00)
− malá disperse chemických posunů (cca. 15 ppm)
+ velká disperse chemických posunů (cca. 210 ppm)
− nízké přirozené zastoupení (1,11 %)
− nízká citlivost (1,76.10−4); po 100% isotopovém obohacení 1,59.10−2
• střední disperse chemických posunů (cca. 30 ppm)
− nízké přirozené zastoupení (0,37 %)
− nízká citlivost (3,85.10−6); po 100% isotopovém obohacení 1,04.10−3
• speciální účely
1H
13C
15N
2H
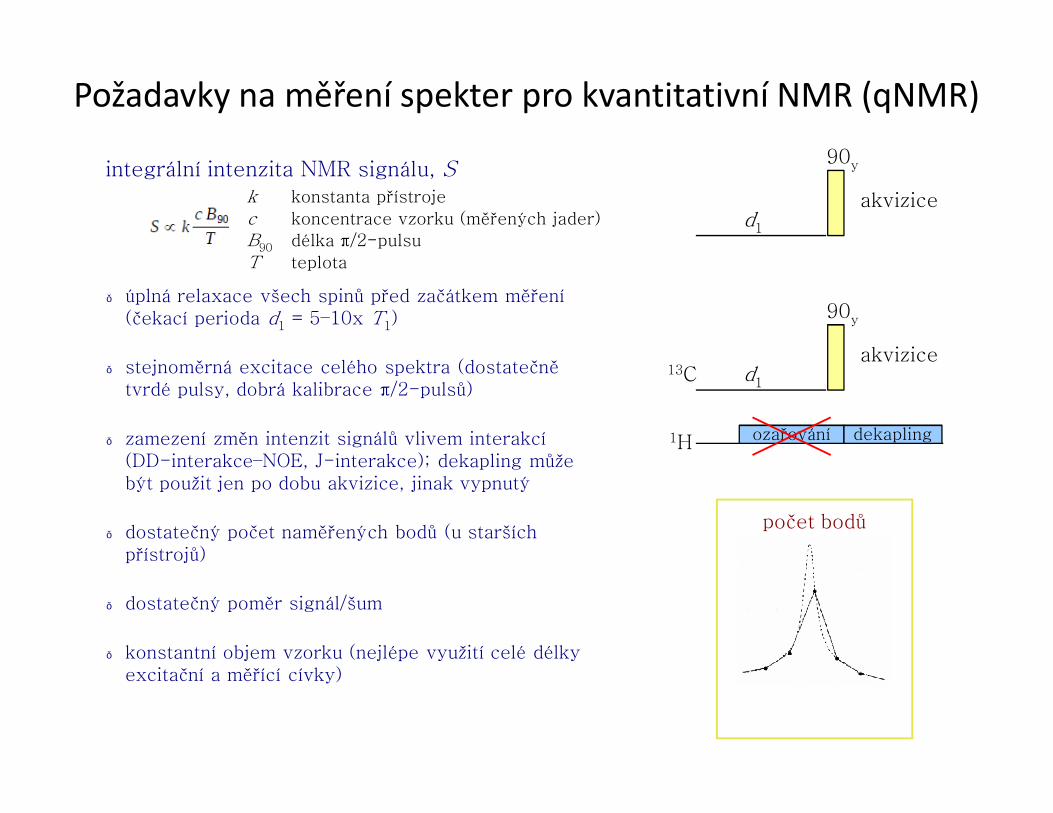
Specifika řešení struktury proteinů pomocí NMR● měření ve fyziologickém prostředí, možnost úpravy fyzikálně-chemických vlastností
prostředí
● sledování průběhu biochemických dějů
● vysoce selektivní odezva na úrovni atomů
Čím jsme omezeni:
● velikost molekuly (ovlivnění T2)
– do 10 kDa (≡10 kg mol−1) [< 70 AA]
□ lze řešit přímo kombinací COSY, TOCSY a NOESY experimentů
– do 20 kDa [< 180 AA]
□ nutné 100% isotopové obohacení 13C a 15N
– do ~100 kDa
□ 100% isotopové obohacení 13C, 15N a částečné nebo úplné obohacení 2H (odstranění 1H jako hlavního zdroje rychlé relaxace 13C)
– větší proteiny
□ přístupný pouze hrubý „náhled“ na celkovou strukturu, sekundární struktura
● koncentrace vzorku
– alespoň 0,2 mM
● dlouhodobá stabilita vzorku
– několik týdnů
Vzorek proteinu pro NMR měření● nutno dosáhnout koncentrace proteinu alespoň 0,2–
0,5 mmol l−1 (obecně více = lépe)
– používá se filtrace přes membrány s mikropóry
(protein zadržen) nebo lyofilizace a opětovné
rozpuštění
● úprava pH pufrem (pH typicky 4–8)
– vyšší pH by způsobilo rychlou výměnu
amidických vodíků s molekulami vody a ztrátu
signálů
● přidání redukčních činidel (R–SH)
– zabránění oxidace cysteinů a následného
vysrážení vzorku
● přidání 5–10 % D2O
– referenční jádro pro spektrometr (lock) roztok vzorku
250–300 µl
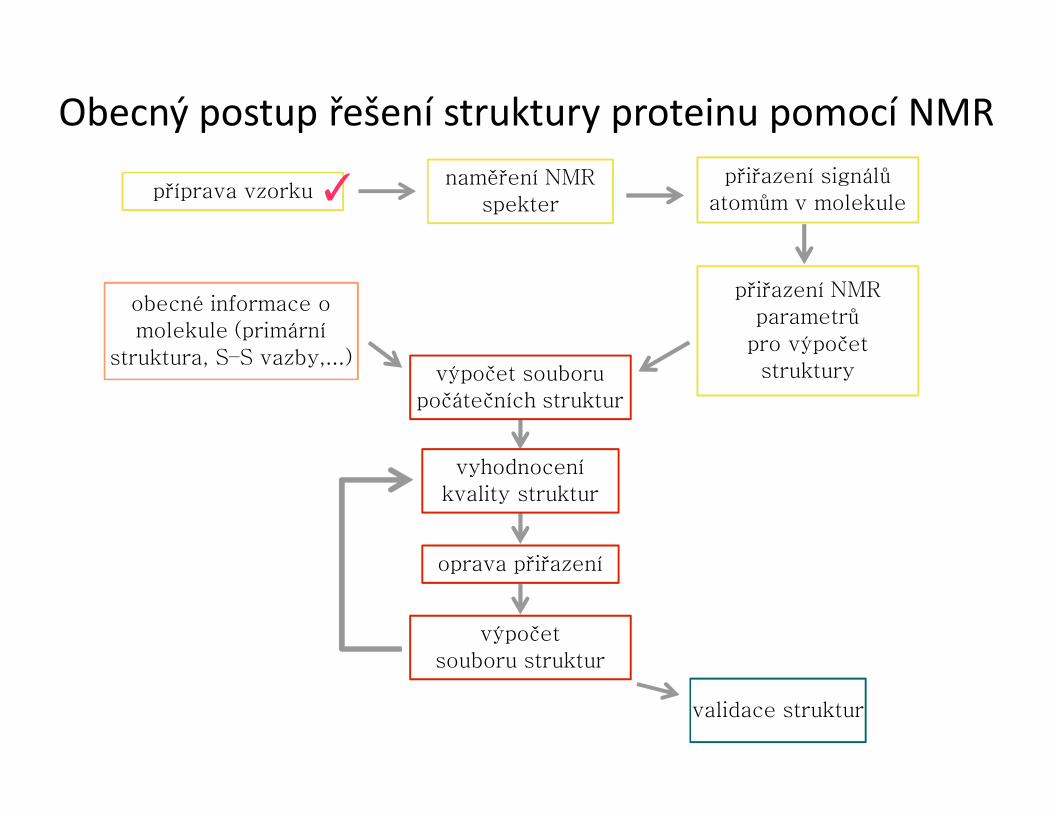
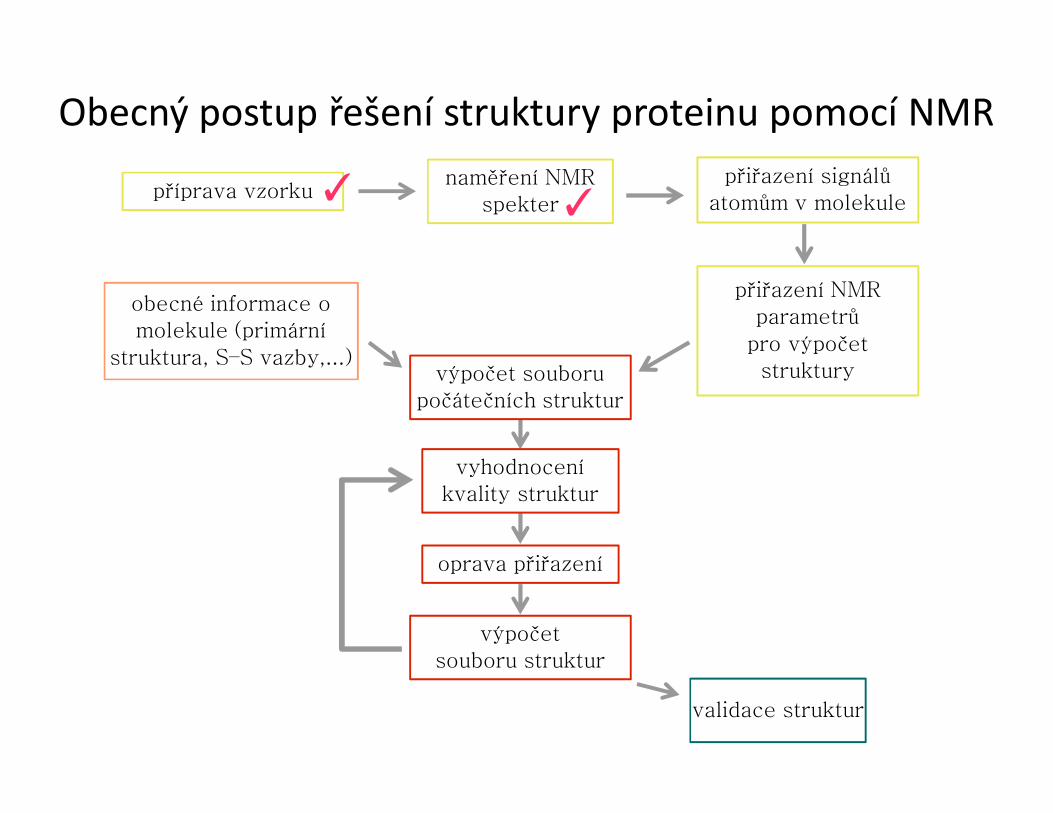
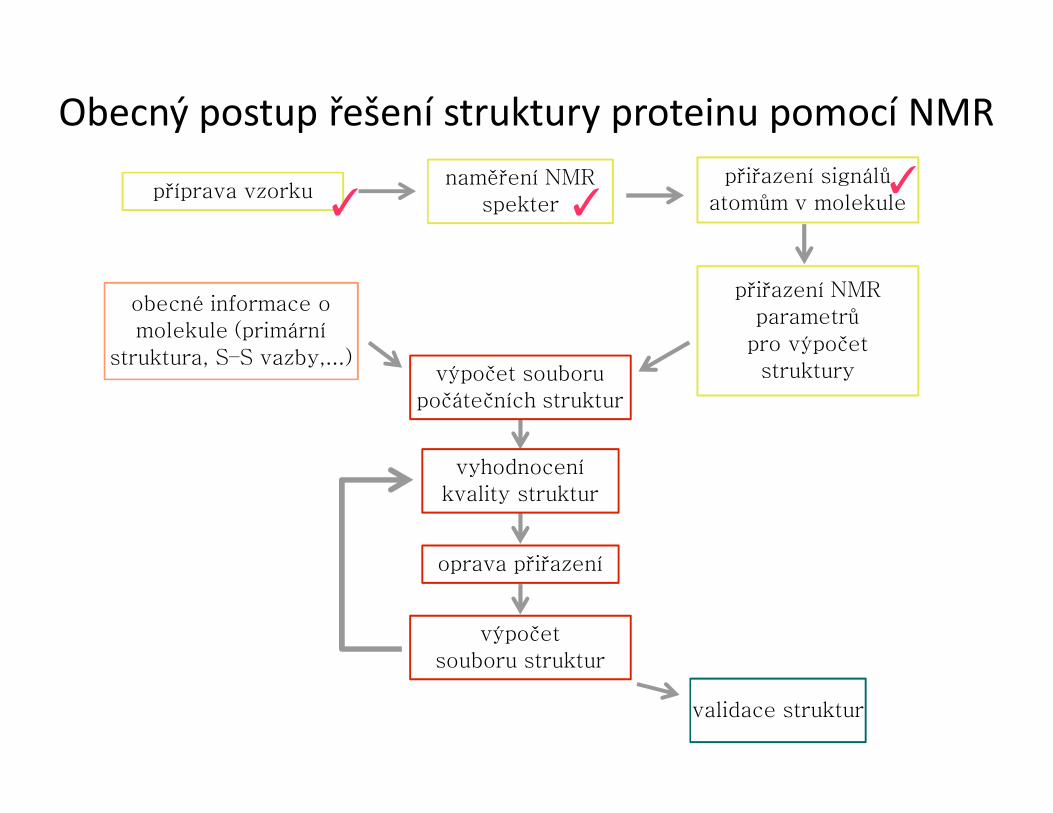
Obecný postup řešení struktury proteinu pomocí NMR
příprava vzorkunaměření NMR
spekter
přiřazení signálů atomům v molekule
přiřazení NMRparametrů
pro výpočetstruktury
obecné informace o molekule (primární
struktura, S–S vazby,...)výpočet souboru
počátečních struktur
vyhodnoceníkvality struktur
oprava přiřazení
výpočetsouboru struktur
validace struktur
✓
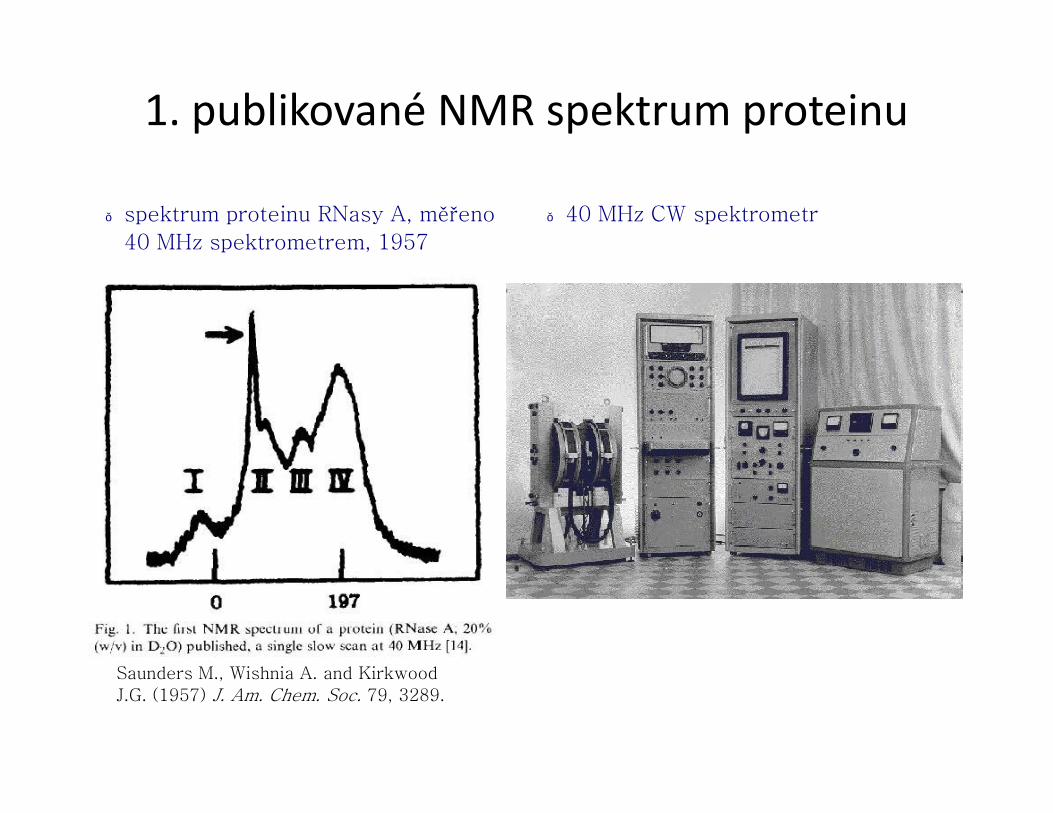
1. publikované NMR spektrum proteinu
● spektrum proteinu RNasy A, měřeno
40 MHz spektrometrem, 1957
● 40 MHz CW spektrometr
Saunders M., Wishnia A. and Kirkwood
J.G. (1957) J. Am. Chem. Soc. 79, 3289.
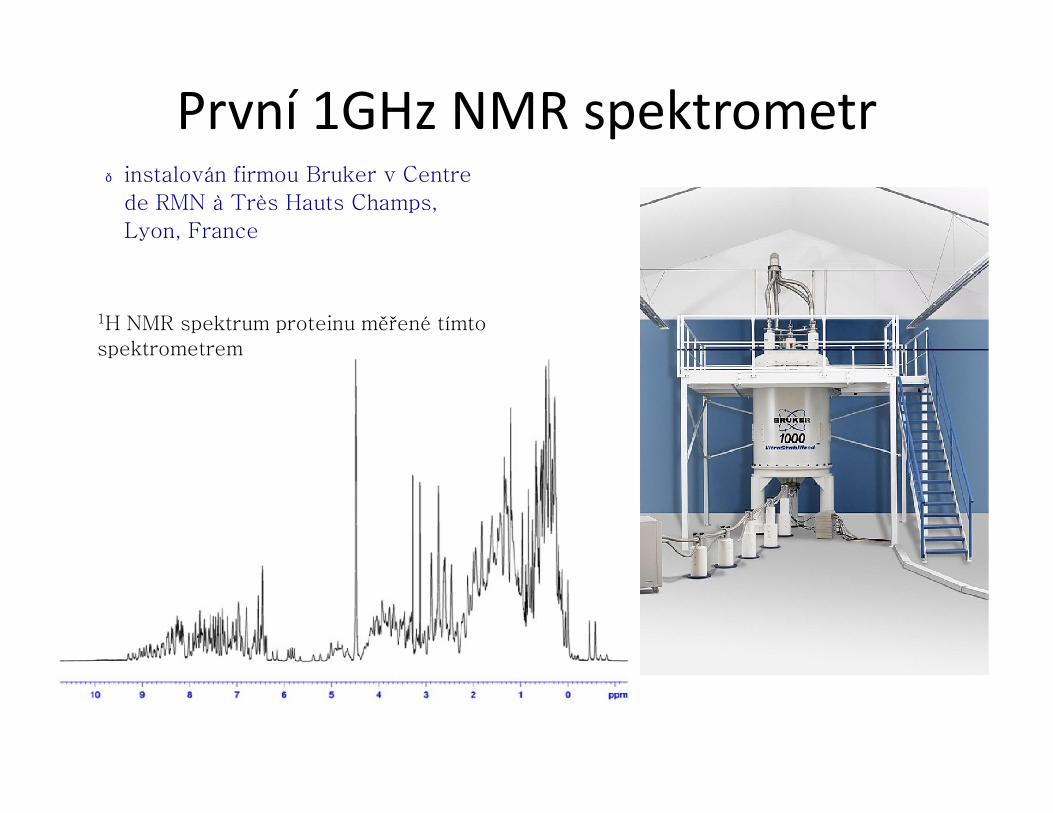
První 1GHz NMR spektrometr● instalován firmou Bruker v Centre
de RMN à Très Hauts Champs,
Lyon, France
1H NMR spektrum proteinu měřené tímto spektrometrem
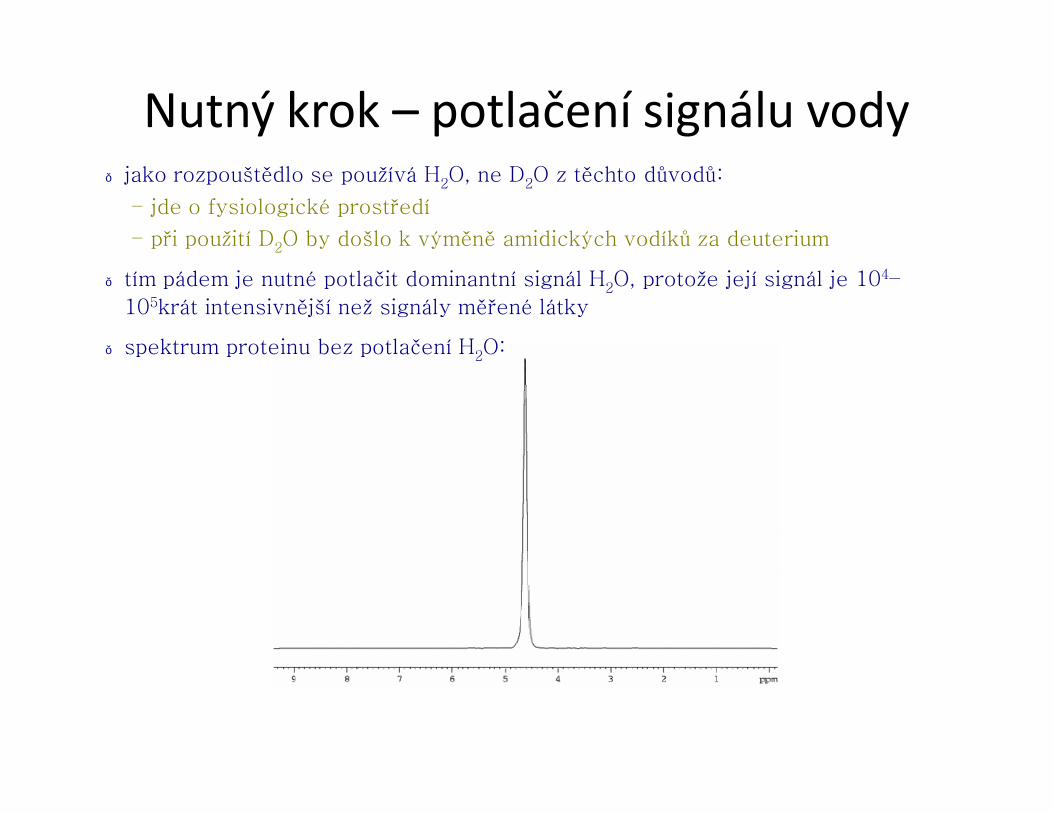
Nutný krok – potlačení signálu vody● jako rozpouštědlo se používá H2O, ne D2O z těchto důvodů:
– jde o fysiologické prostředí
– při použití D2O by došlo k výměně amidických vodíků za deuterium
● tím pádem je nutné potlačit dominantní signál H2O, protože její signál je 104–
105krát intensivnější než signály měřené látky
● spektrum proteinu bez potlačení H2O:
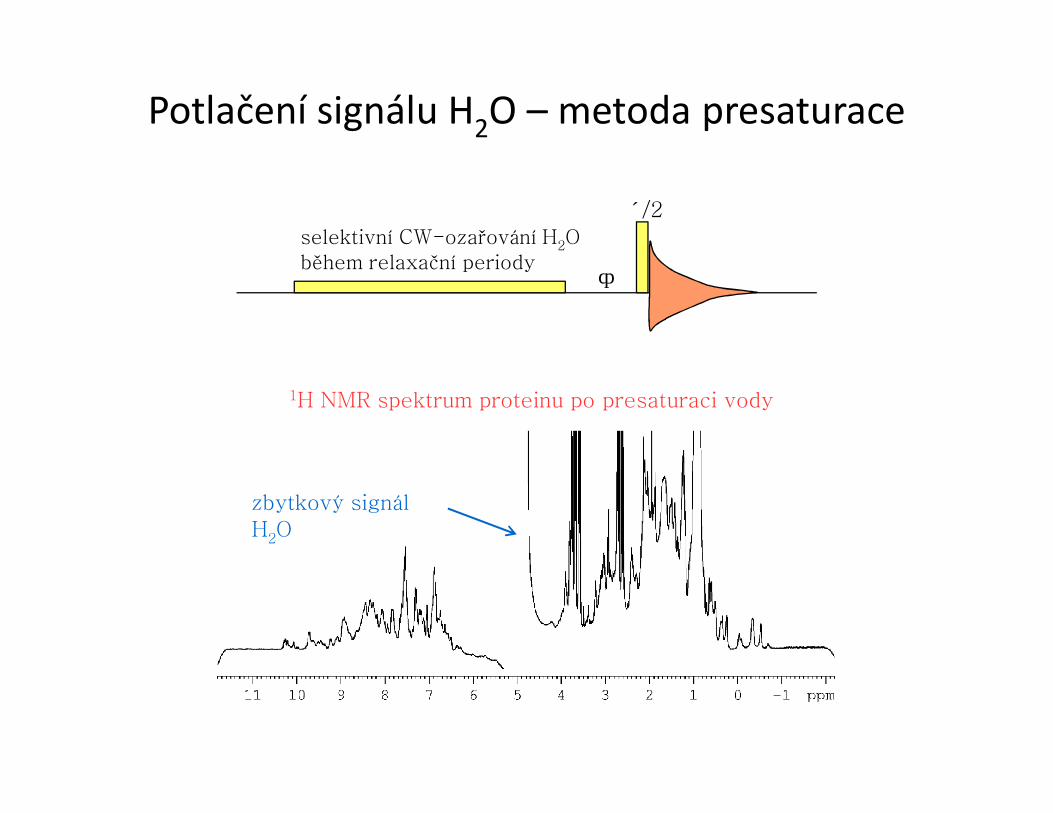
Potlačení signálu H2O – metoda presaturace
selektivní CW-ozařování H2O během relaxační periody
π/2
Δ
zbytkový signál H2O
1H NMR spektrum proteinu po presaturaci vody
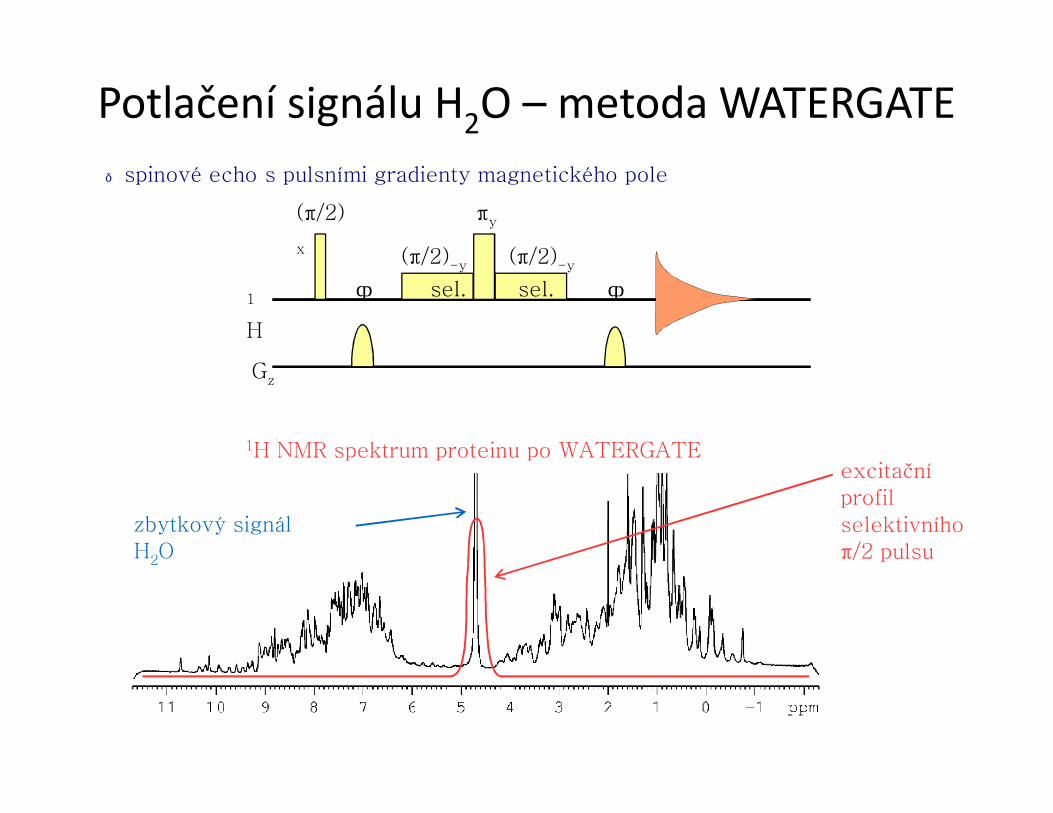
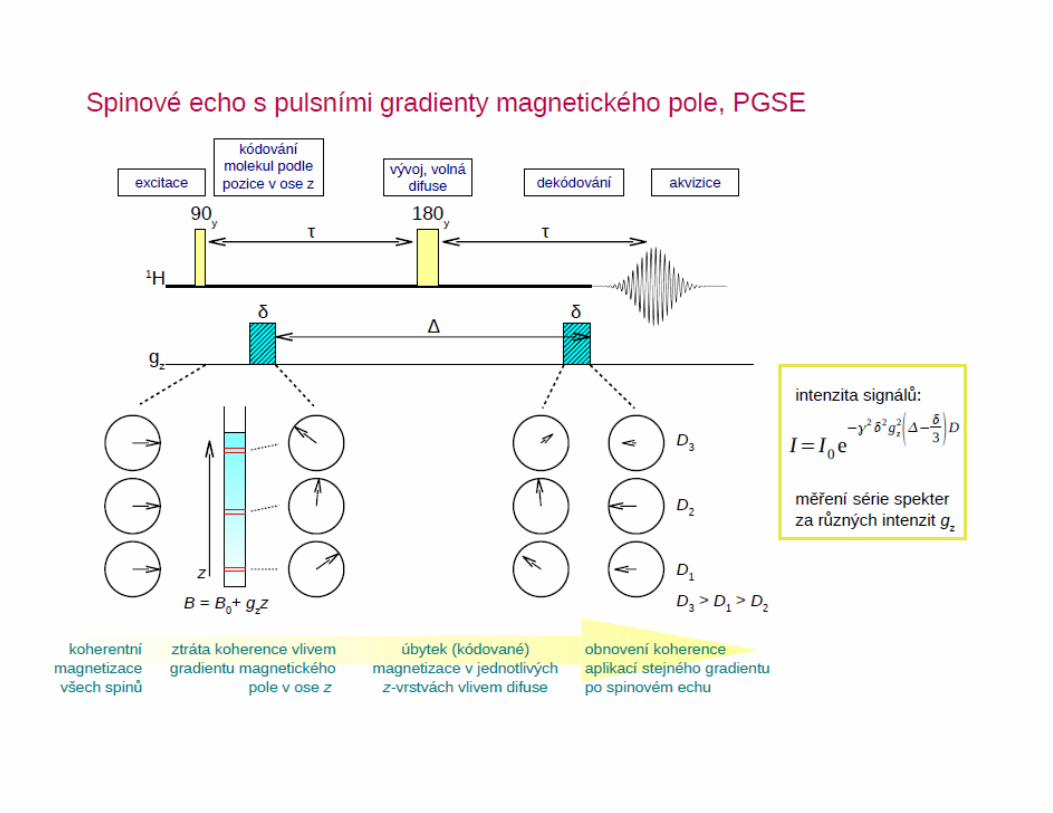
● spinové echo s pulsními gradienty magnetického pole
Potlačení signálu H2O – metoda WATERGATE
(π/2)-y
1
H
Gz
Δ
πy(π/2)
x
sel. sel. Δ(π/2)-y
zbytkový signál H2O
1H NMR spektrum proteinu po WATERGATEexcitační profil selektivníhoπ/2 pulsu
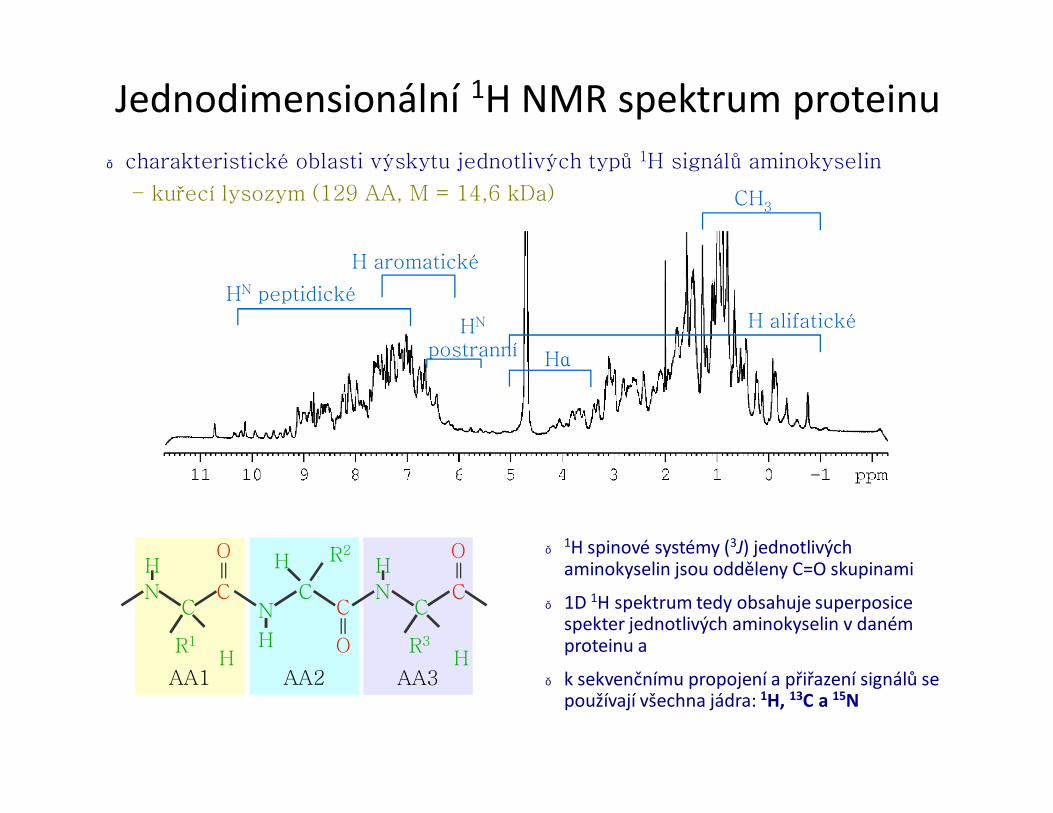
● charakteristické oblasti výskytu jednotlivých typů 1H signálů aminokyselin
– kuřecí lysozym (129 AA, M = 14,6 kDa)
H alifatické
HN peptidické
H aromatické
HN
postranní Hα
CH3
Jednodimensionální 1H NMR spektrum proteinu
AA3AA2AA1
CCNC
NC
C
OH
HH R2
O
HR1
HR3
N
H
C
O
● 1H spinové systémy (3J) jednotlivých aminokyselin jsou odděleny C=O skupinami
● 1D 1H spektrum tedy obsahuje superposice spekter jednotlivých aminokyselin v daném proteinu a
● k sekvenčnímu propojení a přiřazení signálů se používají všechna jádra: 1H, 13C a 15N
Vícedimensionální NMR experimenty● rozlišení informací obsažených v 1D spektrech
– korelace chemických posunů 1H – 13C – 15N
– propojení spinových systémů jednotlivých aminokyselin
– pro dostatečnou citlivost experimentů je nutné použít isotopové obohacení
http://www.protein-nmr.org.uk
1H
(ppm)
1H
(ppm)
15N
(ppm
)
15N
(ppm
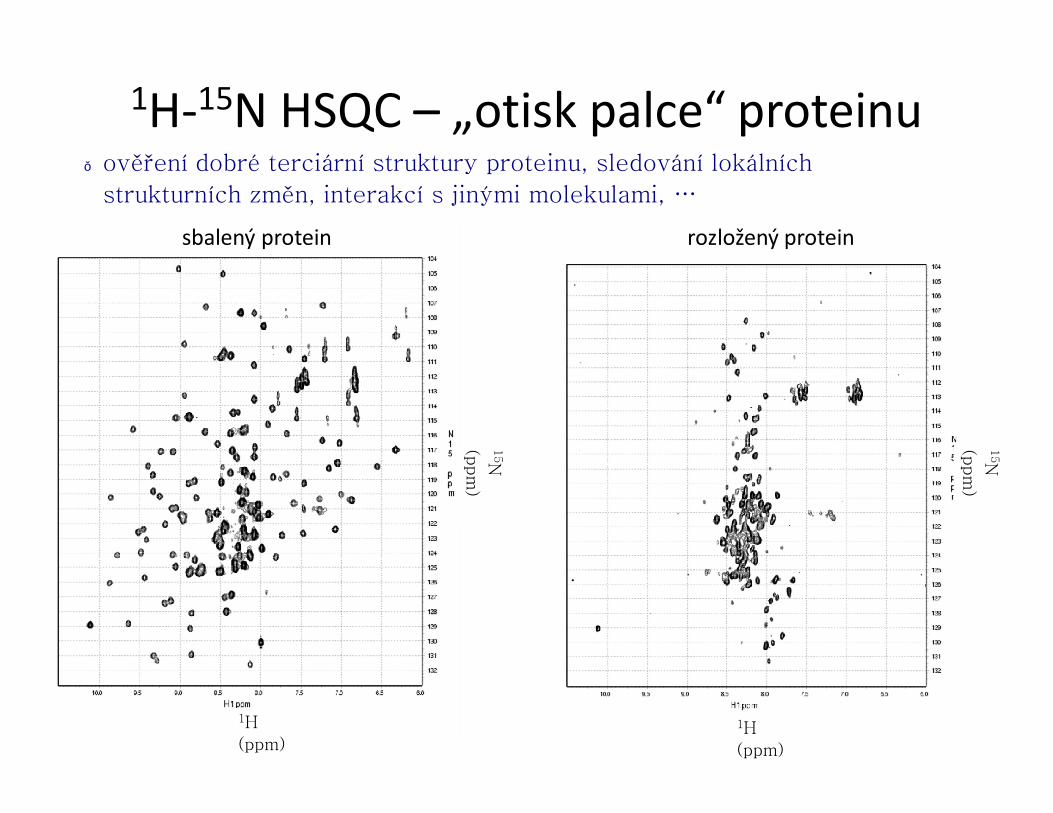
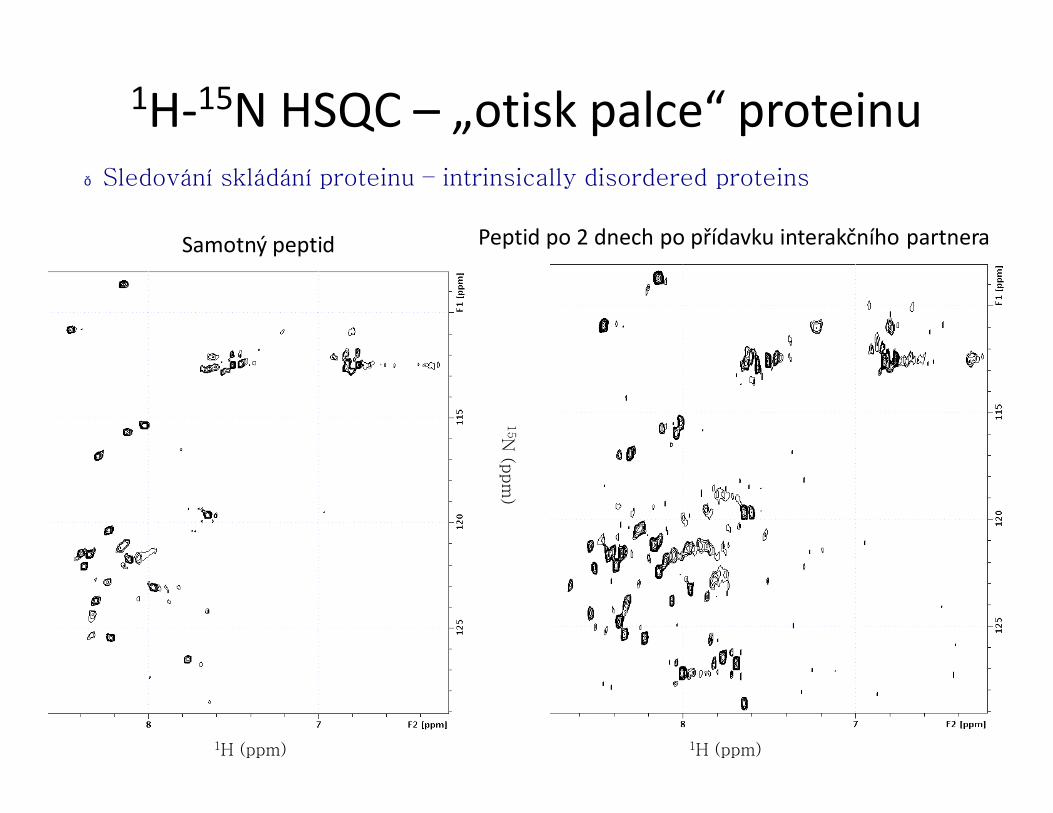
)1H-15N HSQC – „otisk palce“ proteinu
● ověření dobré terciární struktury proteinu, sledování lokálních
strukturních změn, interakcí s jinými molekulami, …
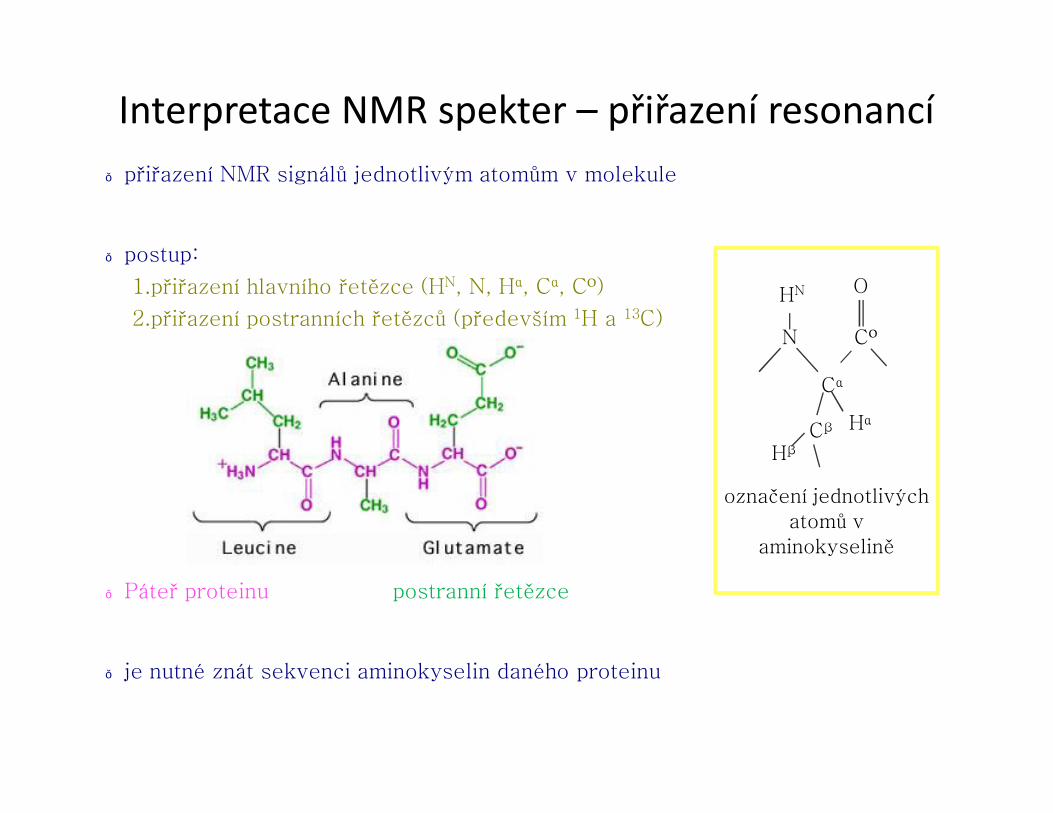
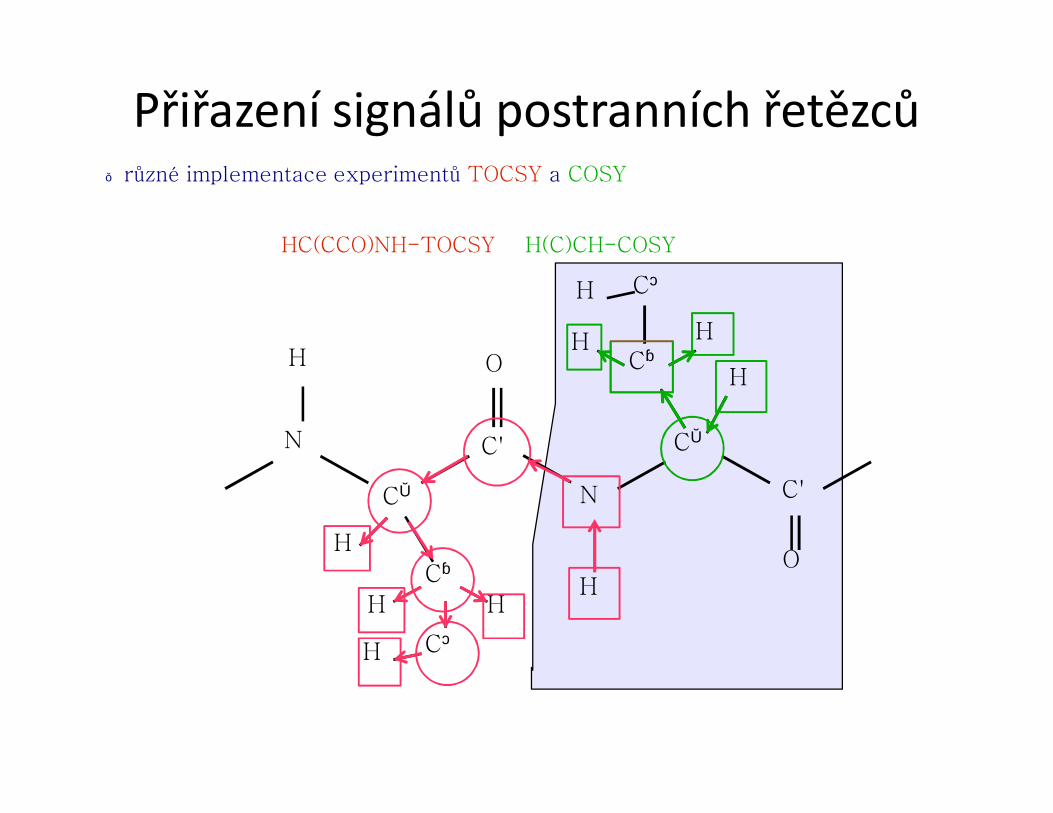
2.přiřazení postranních řetězců (především 1H a 13C)
● Páteř proteinu postranní řetězce
● je nutné znát sekvenci aminokyselin daného proteinu
CO
Cα
O
HαCβ
N
HN
Hβ
označení jednotlivých atomů v
aminokyselině
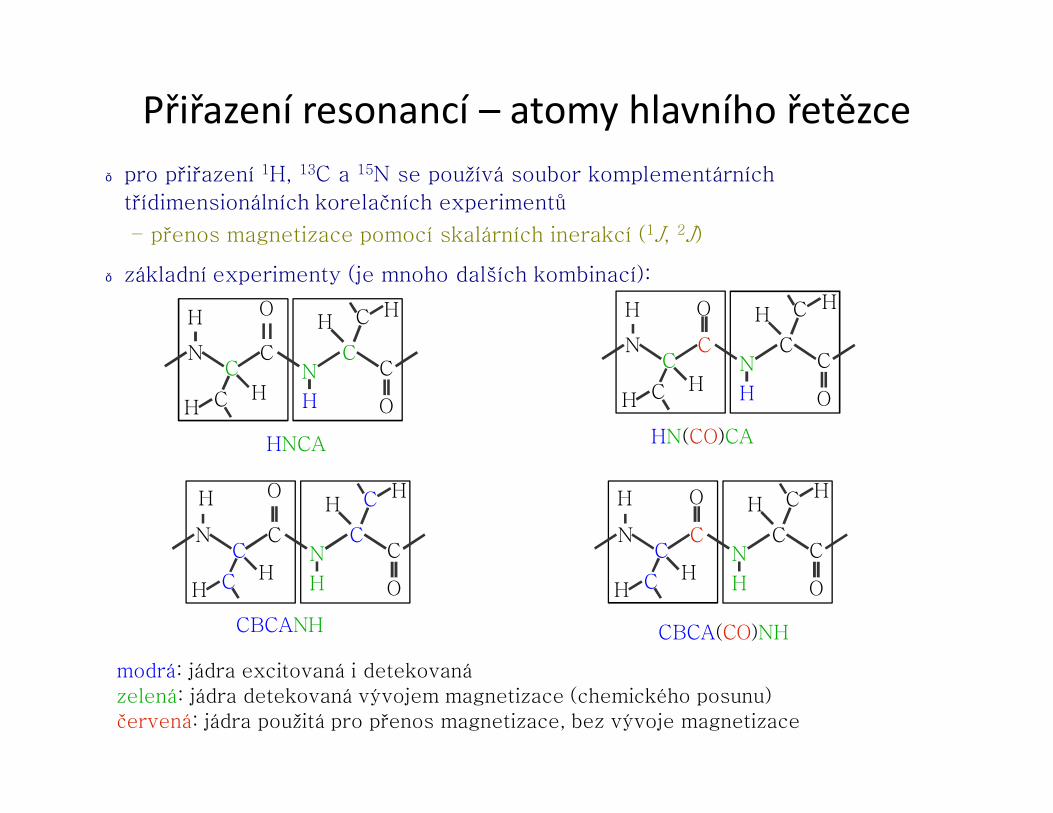
Přiřazení resonancí – atomy hlavního řetězce● pro přiřazení 1H, 13C a 15N se používá soubor komplementárních
třídimensionálních korelačních experimentů
– přenos magnetizace pomocí skalárních inerakcí (1J, 2J)
● základní experimenty (je mnoho dalších kombinací):
CC
NC
C
OH
H C
O
HC
N
H
H
H
HNCA
CC
NC
C
OH
H C
O
H
C
N
H
H
H
HN(CO)CA
CC
NC
C
OH
H CO
H
C
N
H
H
H
CBCANH
CC
NC
C
OH
H C
O
H
C
N
H
H
H
CBCA(CO)NH
modrá: jádra excitovaná i detekovanázelená: jádra detekovaná vývojem magnetizace (chemického posunu)červená: jádra použitá pro přenos magnetizace, bez vývoje magnetizace
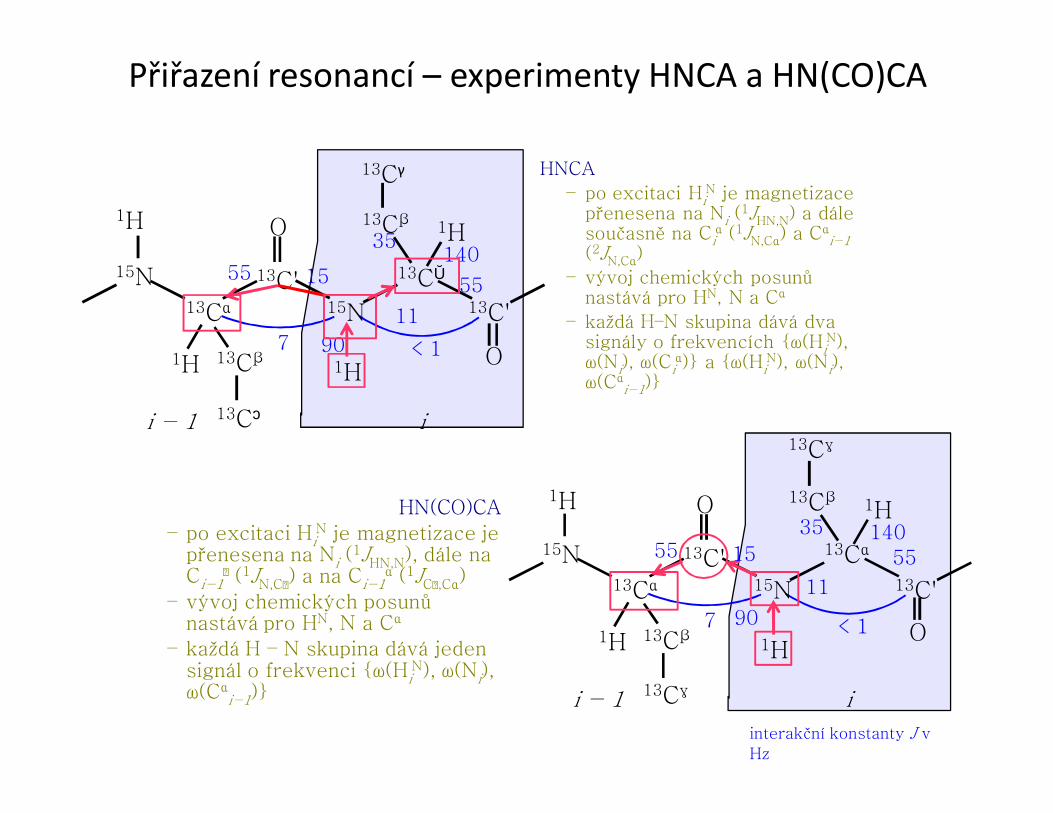
Přiřazení resonancí – experimenty HNCA a HN(CO)CA
13Cα
13C'15N
13Cα
13C'
15N
O
O1H
1H 13Cβ
13Cγ
1H
1H 13Cβ
13Cγ
90
11
15
7
55140
55
35
i − 1 i
< 1
13Cα
13C'15N
13Cα
13C'
15N
O
O1H
1H 13Cβ
13Cγ
1H
1H 13Cβ
13Cγ
90
11
15
7
55140
55
35
i − 1 i
< 1
interakční konstanty J v
Hz
HN(CO)CA
– po excitaci HiN je magnetizace je
přenesena na Ni (1JHN,N), dále na Ci−1
(1JN,C) a na Ci−1α (1JC,Cα)
– vývoj chemických posunů nastává pro HN, N a Cα
– každá H – N skupina dává jeden signál o frekvenci {ω(Hi
N), ω(Ni), ω(Cα
i−1)}
HNCA
– po excitaci HiN je magnetizace
přenesena na Ni (1JHN,N) a dále současně na Ci
α (1JN,Cα) a Cαi−1
(2JN,Cα)
– vývoj chemických posunů nastává pro HN, N a Cα
– každá H–N skupina dává dva signály o frekvencích {ω(Hi
N), ω(Ni), ω(Ci
α)} a {ω(HiN), ω(Ni),
ω(Cαi−1)}
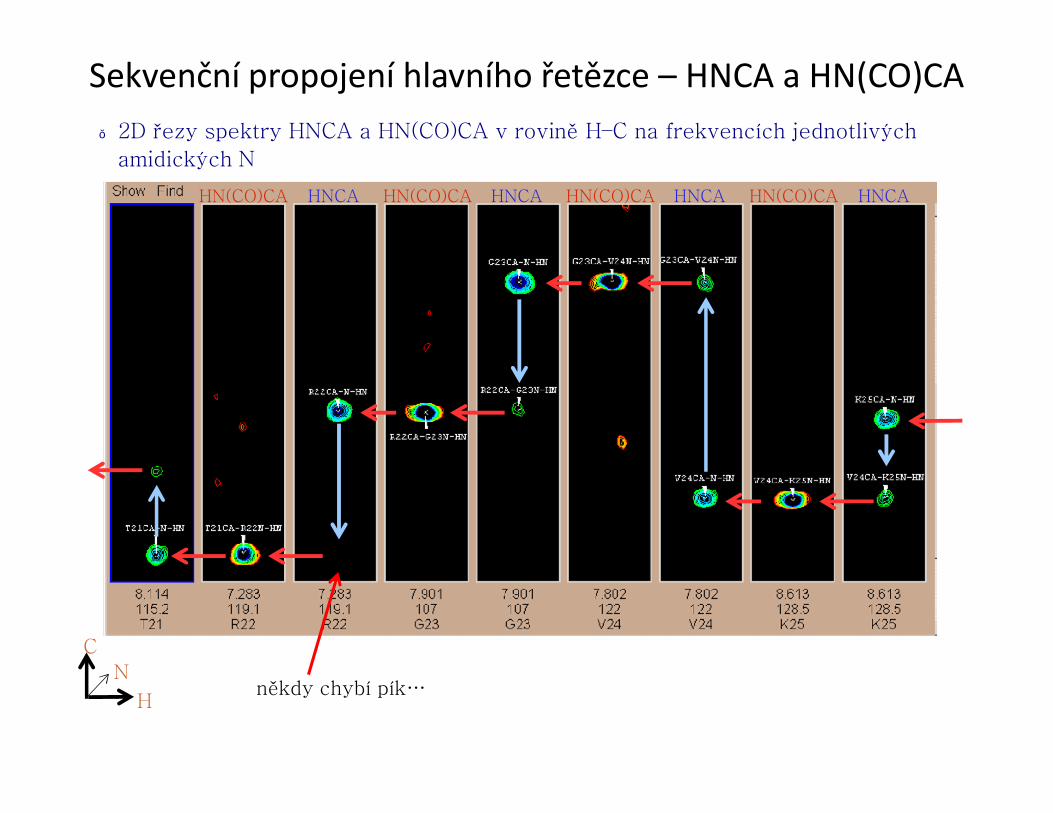
Sekvenční propojení hlavního řetězce – HNCA a HN(CO)CA● 2D řezy spektry HNCA a HN(CO)CA v rovině H–C na frekvencích jednotlivých
Nukleární Overhauserův efekt (NOE)● přímá dipól-dipólová interakce mezi atomy I a S vlivem křížové relaxace
● rychlostní konstanta křížové relaxace mezi dvěma jádry 1H, σIS:
γH ... gyromagnetická konstanta 1H
τc ... korelační čas molekulyω ... Larmorova frekvencerIS ... vzdálenost interagujících jader
H H
dosah interakce do 5–6 Å
●NOE je hlavní zdroj informace o struktuře proteinu. Cílem je nalézt co největší počet NOE interakcí a jednoznačně je přiřadit dvojicím konkrétních vodíkových atomů v molekule.●Pravidelné prvky sekundární struktury tvoří charakteristické sítě NOE kontaktů.●Z NOE se nepočítají přesné vzdálenosti vodíků, ale rozdělí se do pásem podle intenzity.●Např. (1,8–2,5) Å; (1,8–3,5) Å; (1,8–5) Å.
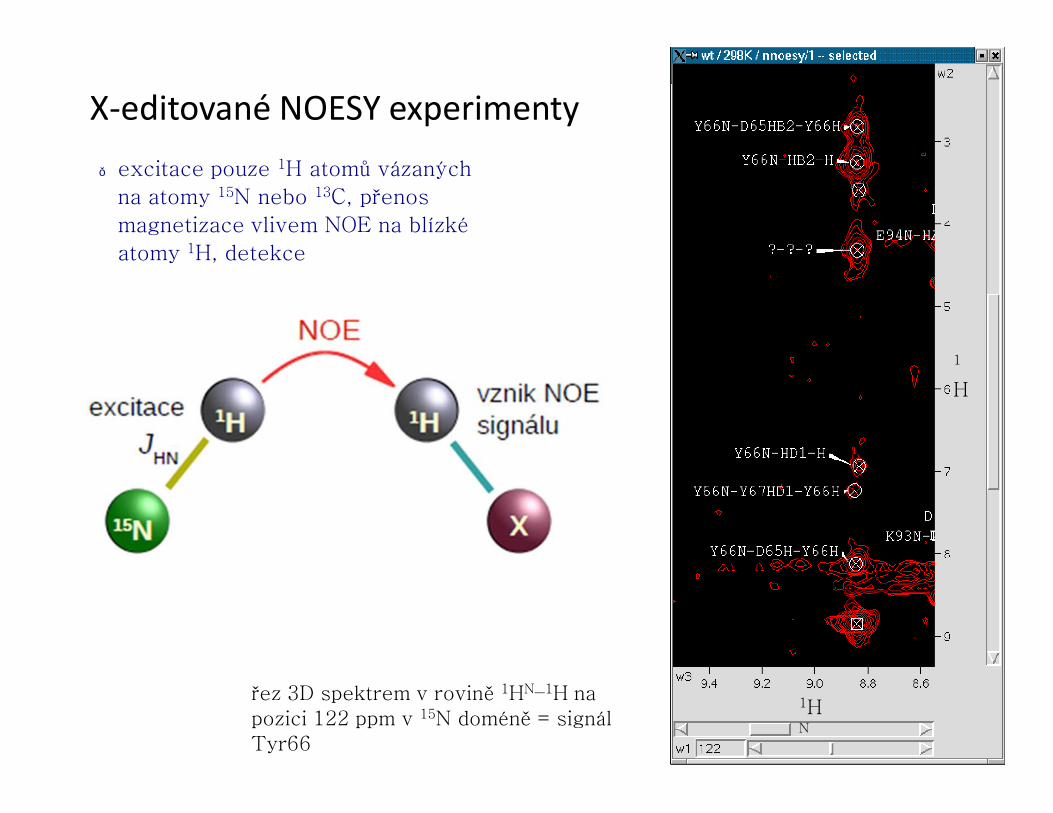
X-editované NOESY experimenty● excitace pouze 1H atomů vázaných
na atomy 15N nebo 13C, přenos
magnetizace vlivem NOE na blízké
atomy 1H, detekce
1HN
1
H
řez 3D spektrem v rovině 1HN–1H na pozici 122 ppm v 15N doméně = signál Tyr66
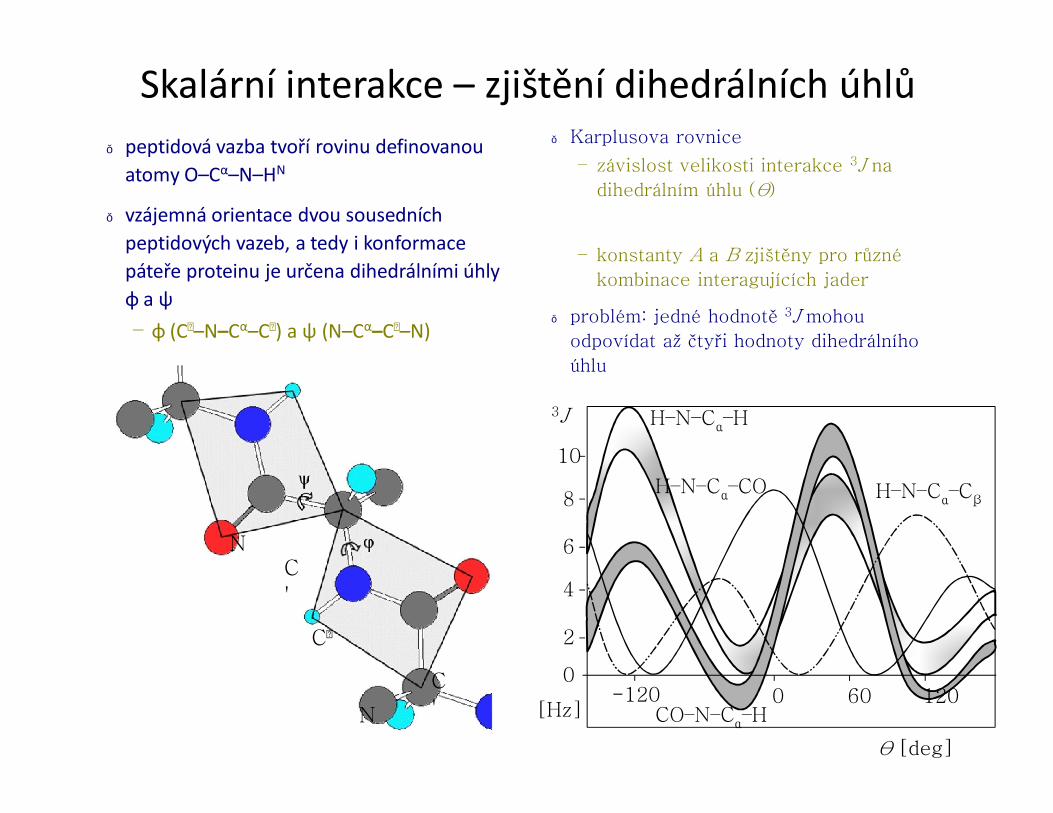
● vzájemná orientace dvou sousedních peptidových vazeb, a tedy i konformacepáteře proteinu je určena dihedrálními úhly φ a ψ– φ (C–N–Cα–C) a ψ (N–Cα–C–N)
● Karplusova rovnice
– závislost velikosti interakce 3J na
dihedrálním úhlu (θ)
– konstanty A a B zjištěny pro různé
kombinace interagujících jader
● problém: jedné hodnotě 3J mohou
odpovídat až čtyři hodnoty dihedrálního
úhlu
N
C'
C
N
C'
3J
[Hz]
10
8
6
4
2
0-120 600 120
H–N–Cα–H
CO–N–Cα–H
H–N–Cα–CβH–N–Cα–CO
θ [deg]
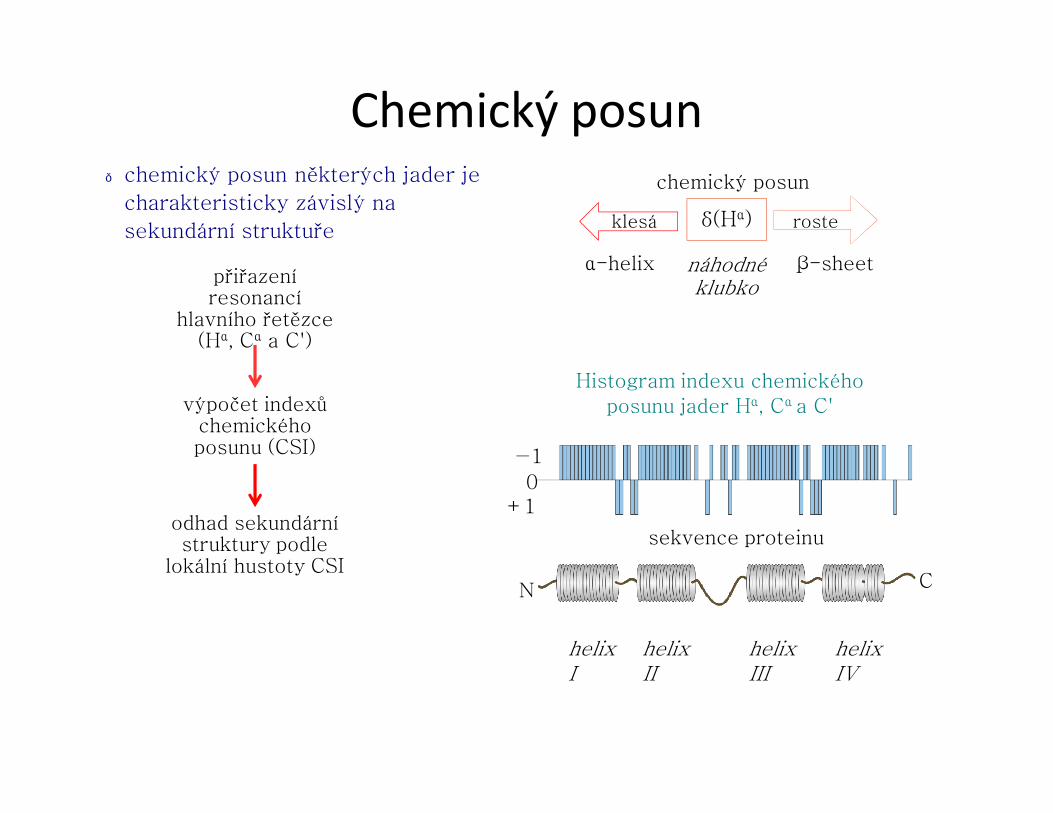
Chemický posun● chemický posun některých jader je
charakteristicky závislý na
sekundární struktuře
přiřazení resonancí
hlavního řetězce(Hα, Cα a C')
výpočet indexů chemického posunu (CSI)
odhad sekundární struktury podle
lokální hustoty CSI
náhodnéklubko
δ(Hα)
α-helix β-sheet
klesá roste
chemický posun
Histogram indexu chemického posunu jader Hα, Cα a C'
CN
−1
0+1
sekvence proteinu
helixI
helixII
helixIII
helixIV
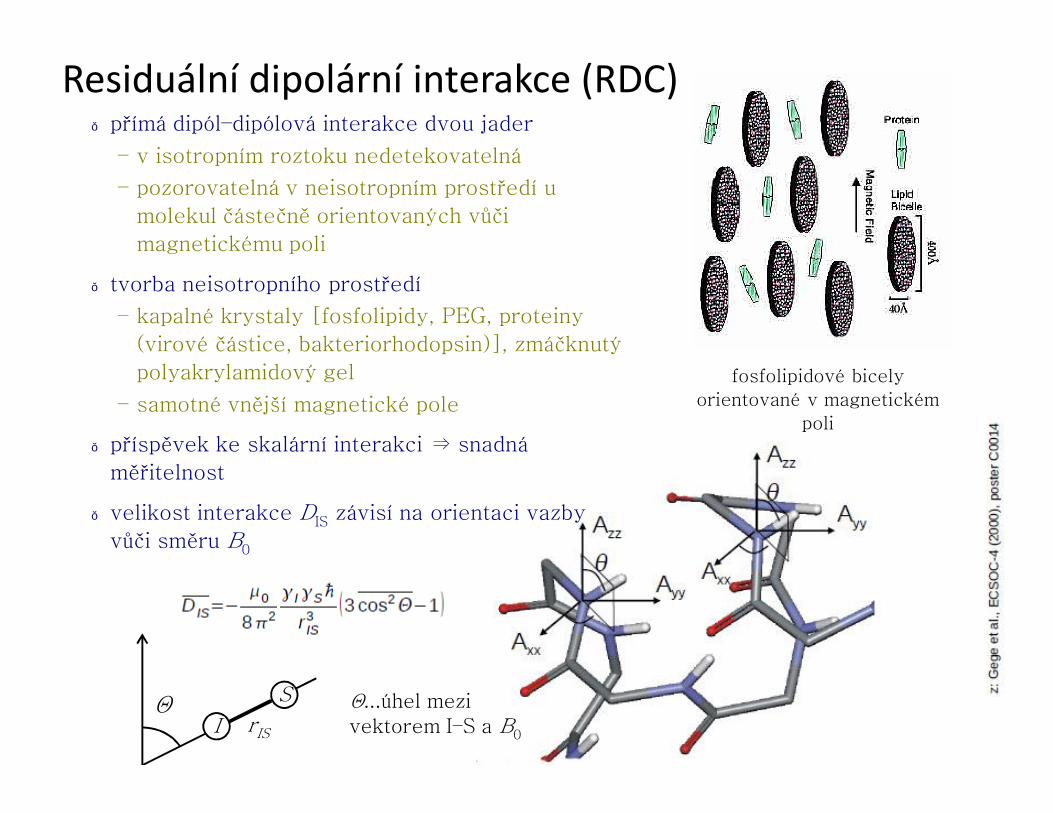
Residuální dipolární interakce (RDC)● přímá dipól–dipólová interakce dvou jader
– v isotropním roztoku nedetekovatelná
– pozorovatelná v neisotropním prostředí u
molekul částečně orientovaných vůči
magnetickému poli
● tvorba neisotropního prostředí
– kapalné krystaly [fosfolipidy, PEG, proteiny
(virové částice, bakteriorhodopsin)], zmáčknutý
polyakrylamidový gel
– samotné vnější magnetické pole
● příspěvek ke skalární interakci ⇒ snadná
měřitelnost
● velikost interakce DIS závisí na orientaci vazby
vůči směru B0
I
S
rIS
Θ Θ...úhel mezi vektorem I–S a B0
fosfolipidové bicely
orientované v magnetickém
poli
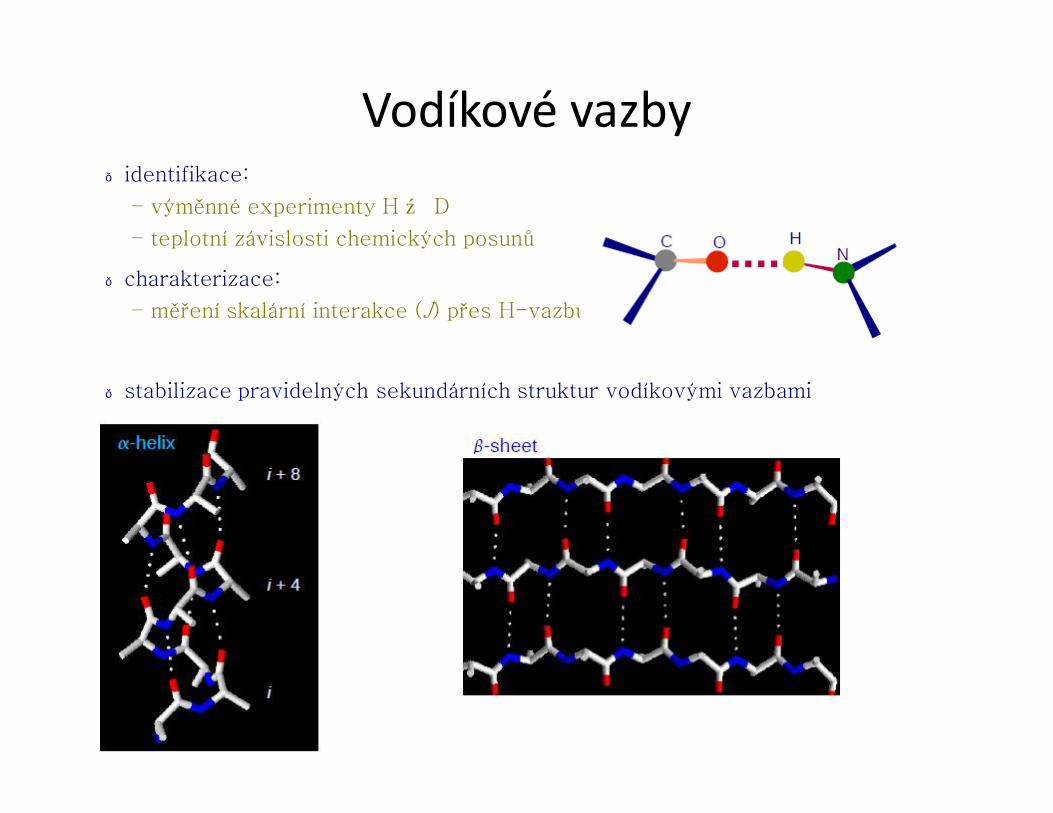
Vodíkové vazby● identifikace:
– výměnné experimenty H ↔ D
– teplotní závislosti chemických posunů
● charakterizace:
– měření skalární interakce (J) přes H-vazbu
● stabilizace pravidelných sekundárních struktur vodíkovými vazbami
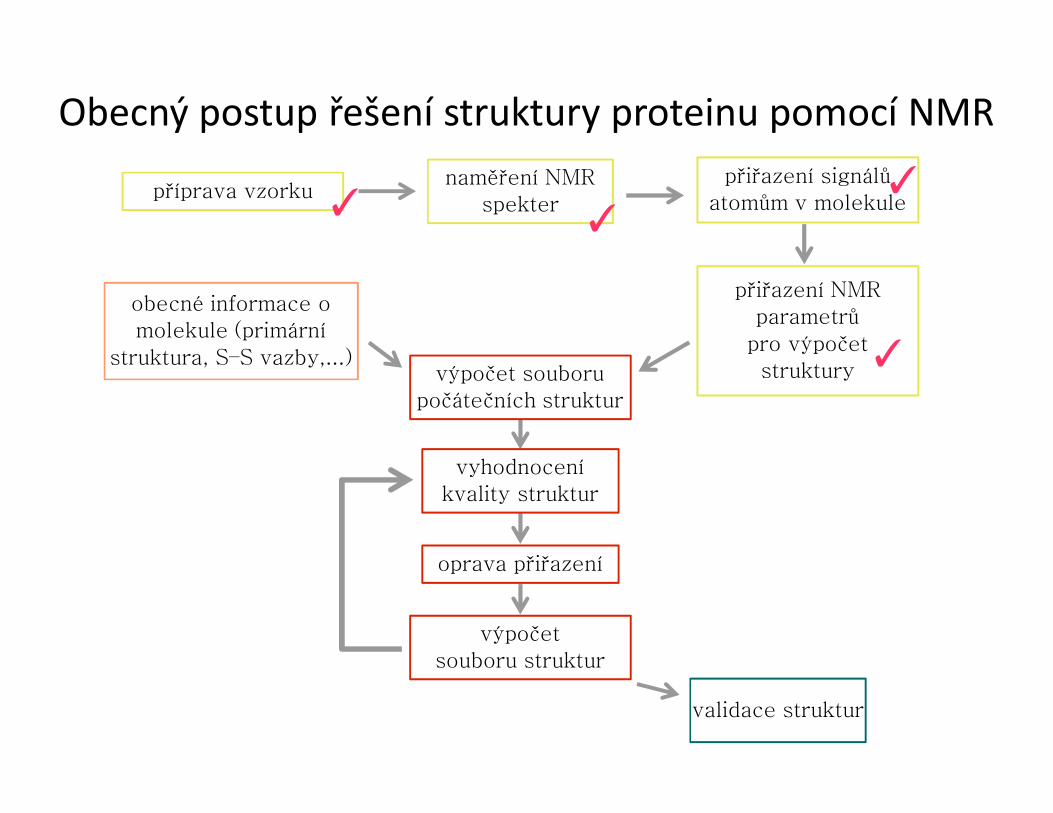
Obecný postup řešení struktury proteinu pomocí NMR
příprava vzorkunaměření NMR
spekter
přiřazení signálů atomům v molekule
přiřazení NMRparametrů
pro výpočetstruktury
obecné informace o molekule (primární
struktura, S–S vazby,...)výpočet souboru
počátečních struktur
vyhodnoceníkvality struktur
oprava přiřazení
výpočetsouboru struktur
validace struktur
✓✓
✓
✓
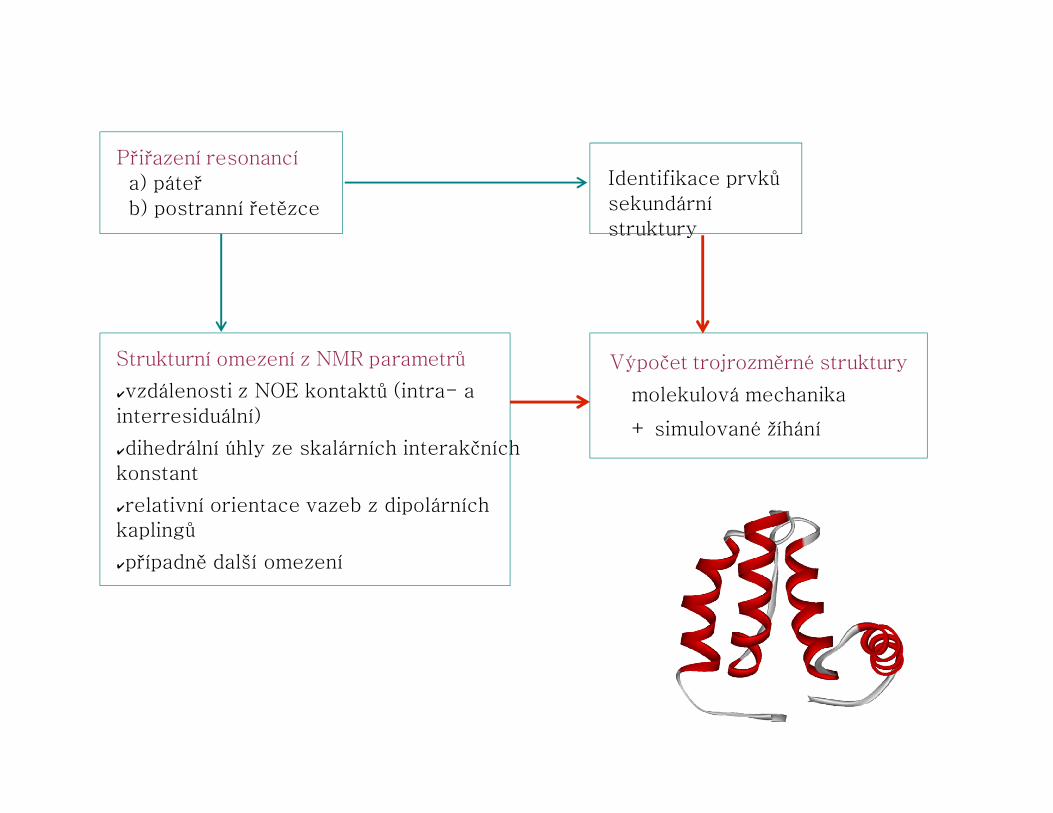
Přiřazení resonancía) páteřb) postranní řetězce
Strukturní omezení z NMR parametrů
✔vzdálenosti z NOE kontaktů (intra- a interresiduální)
✔dihedrální úhly ze skalárních interakčních konstant
✔relativní orientace vazeb z dipolárníchkaplingů
✔případně další omezení
Výpočet trojrozměrné struktury
molekulová mechanika
+ simulované žíhání
Identifikace prvků sekundární struktury
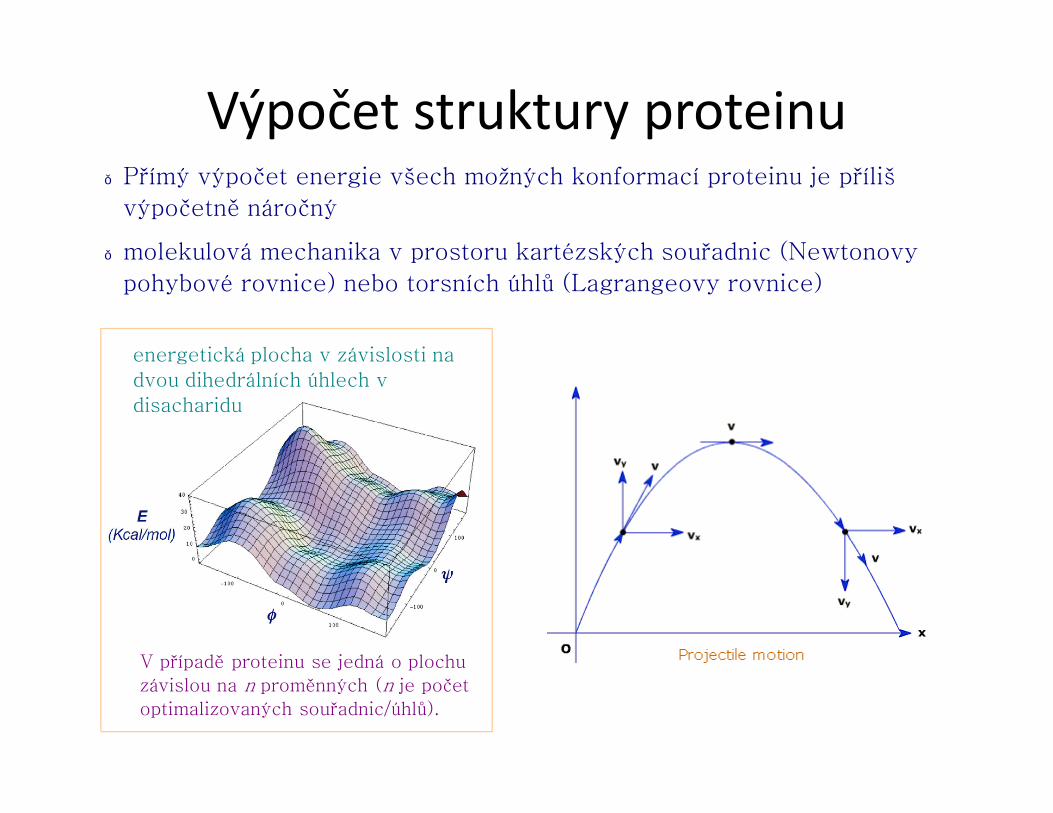
Výpočet struktury proteinu● Přímý výpočet energie všech možných konformací proteinu je příliš
výpočetně náročný
● molekulová mechanika v prostoru kartézských souřadnic (Newtonovy
pohybové rovnice) nebo torsních úhlů (Lagrangeovy rovnice)
energetická plocha v závislosti na dvou dihedrálních úhlech v disacharidu
V případě proteinu se jedná o plochu
závislou na n proměnných (n je počet
optimalizovaných souřadnic/úhlů).
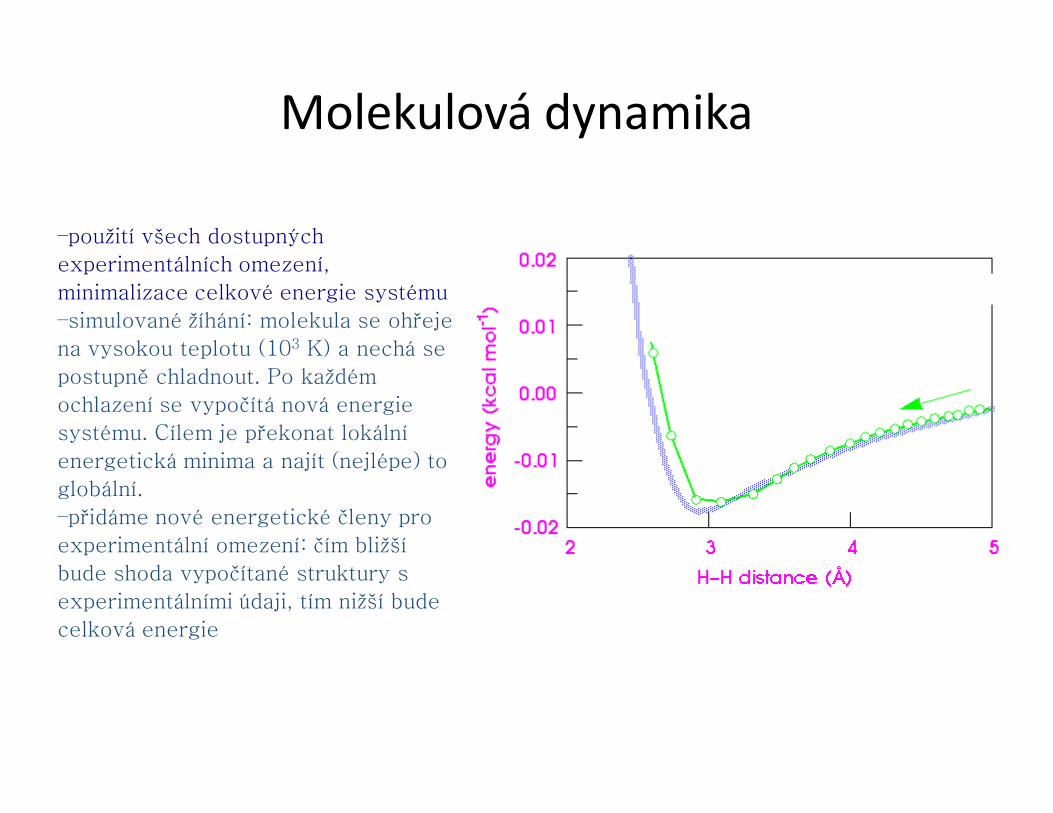
–použití všech dostupných
experimentálních omezení,
minimalizace celkové energie systému
–simulované žíhání: molekula se ohřeje
na vysokou teplotu (103 K) a nechá se
postupně chladnout. Po každém
ochlazení se vypočítá nová energie
systému. Cílem je překonat lokální
energetická minima a najít (nejlépe) to
globální.
–přidáme nové energetické členy pro
experimentální omezení: čím bližší
bude shoda vypočítané struktury s
experimentálními údaji, tím nižší bude
celková energie
Molekulová dynamika
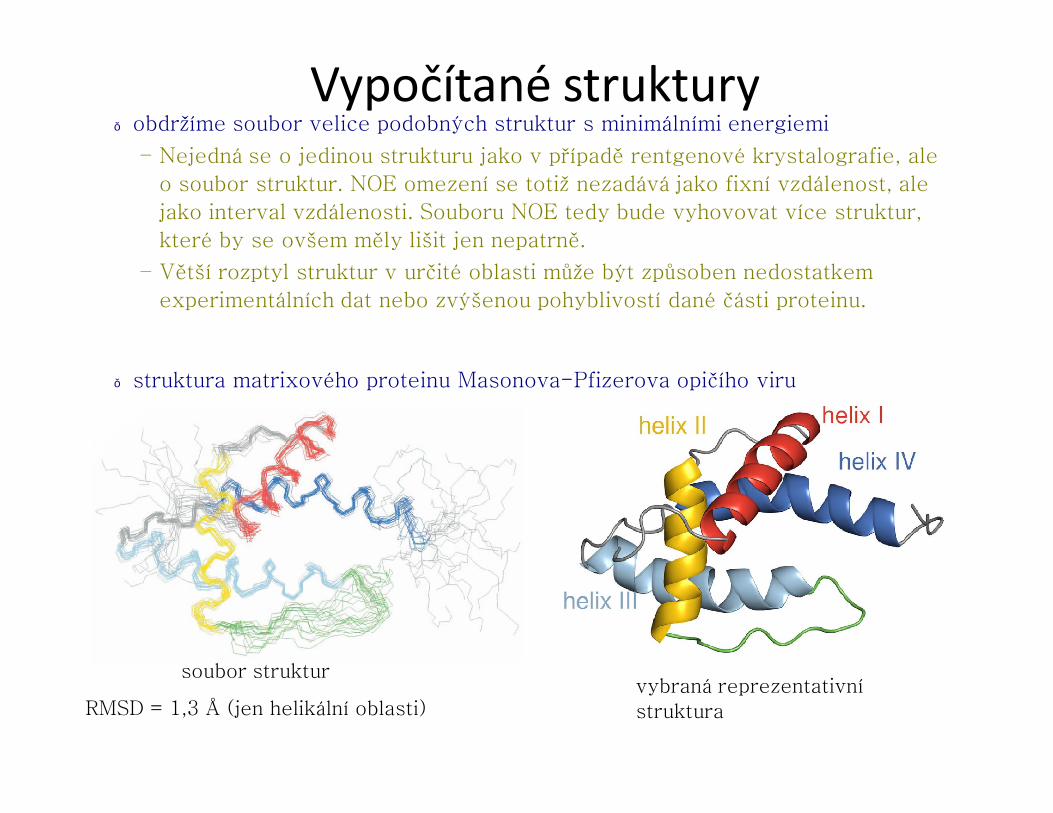
Vypočítané struktury● obdržíme soubor velice podobných struktur s minimálními energiemi
– Nejedná se o jedinou strukturu jako v případě rentgenové krystalografie, ale
o soubor struktur. NOE omezení se totiž nezadává jako fixní vzdálenost, ale
jako interval vzdálenosti. Souboru NOE tedy bude vyhovovat více struktur,
které by se ovšem měly lišit jen nepatrně.
– Větší rozptyl struktur v určité oblasti může být způsoben nedostatkem
experimentálních dat nebo zvýšenou pohyblivostí dané části proteinu.
● struktura matrixového proteinu Masonova-Pfizerova opičího viru
soubor strukturvybraná reprezentativní strukturaRMSD = 1,3 Å (jen helikální oblasti)
Interakce proteinu s ligandem• Protein-protein, protein-NK, protein-malá molekula, oligomerace• Interaguje specificky?• Kde interaguje?• Síla interakce (vhodné i pro slabší interakci)
• Farmaceutický průmysl• Proteomické studie
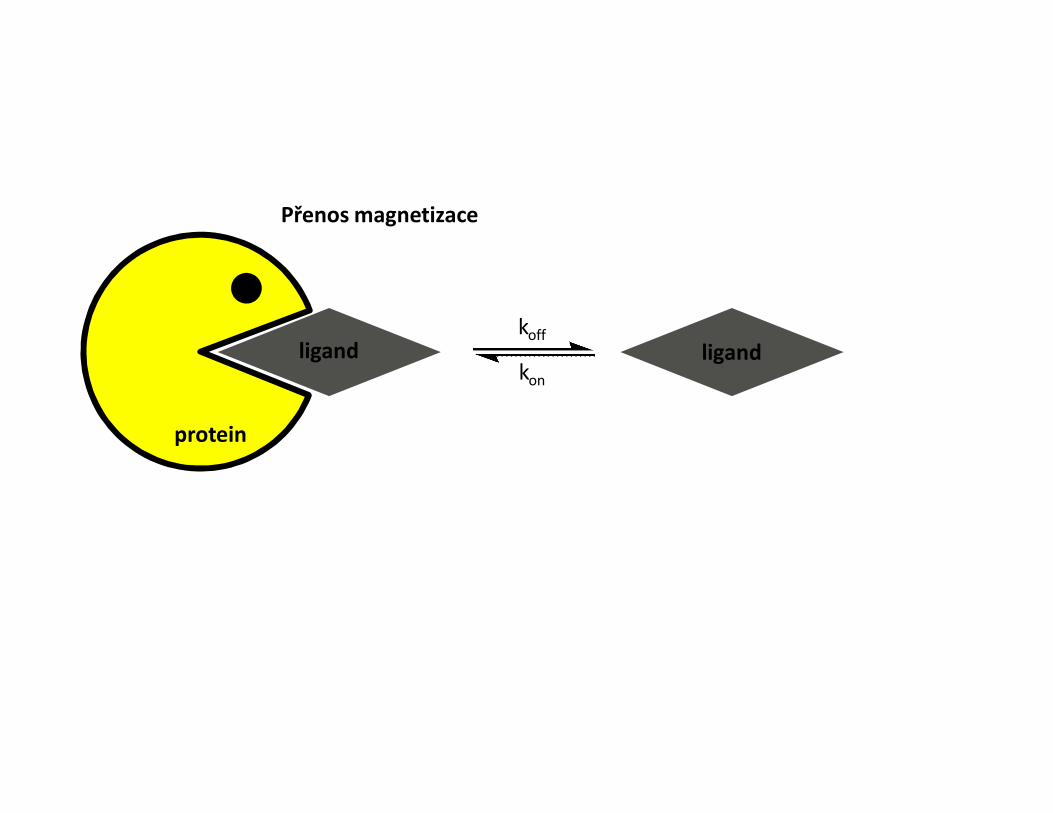
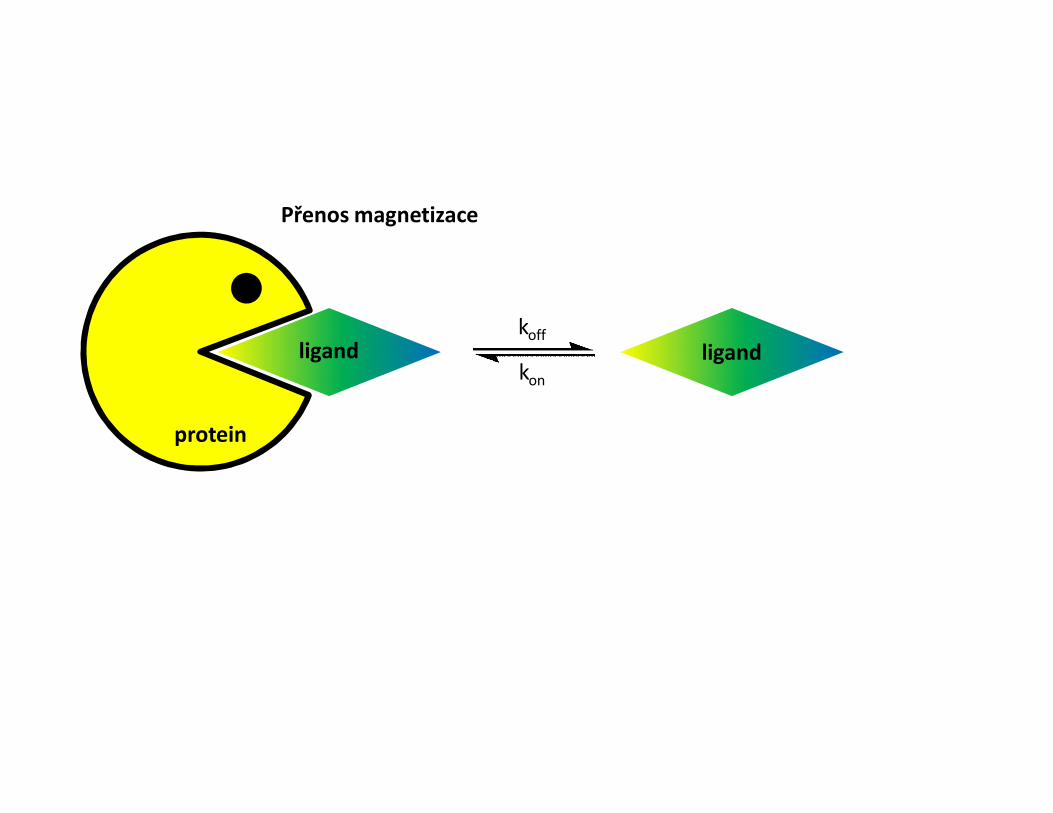
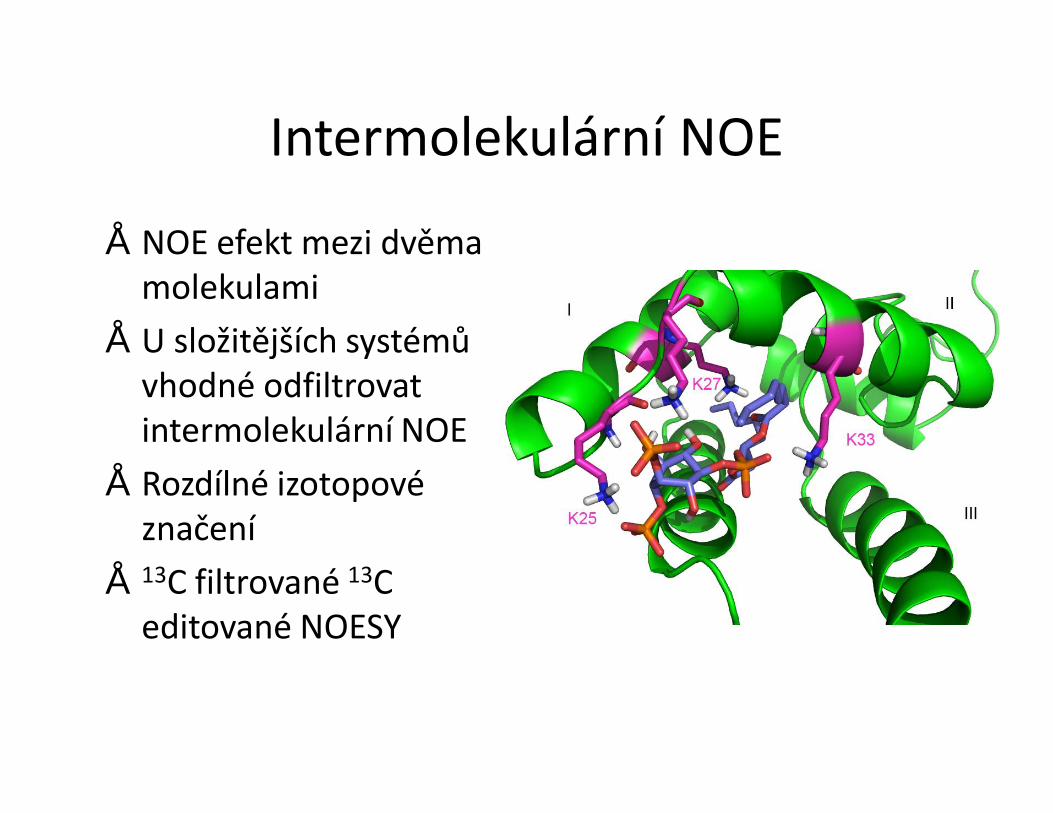
• Metody pro měření interakce pomocí NMR• Sledování změn chemických posunů• Intermolekulární NOE• Transferred NOESY• Mapování H-D chemické výměny NH skupin• Mapování pomocí paramagnetické látky• Sledování změny dynamiky proteinu
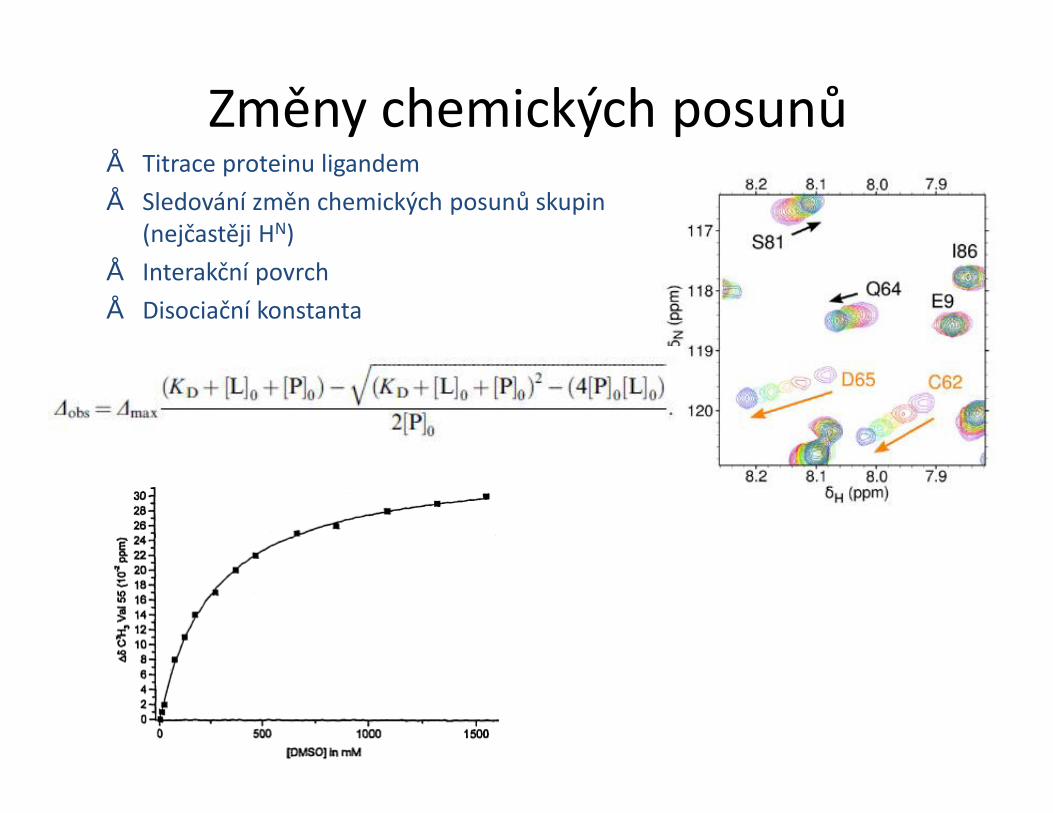
Změny chemických posunů• Titrace proteinu ligandem• Sledování změn chemických posunů skupin
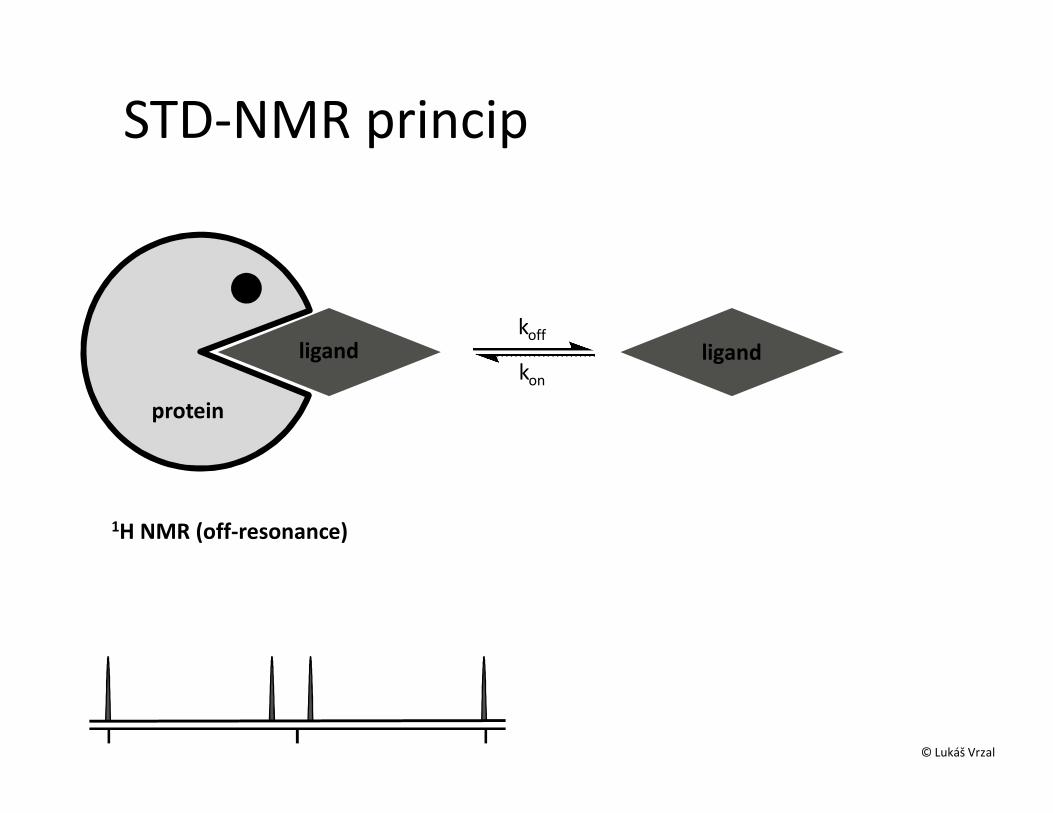
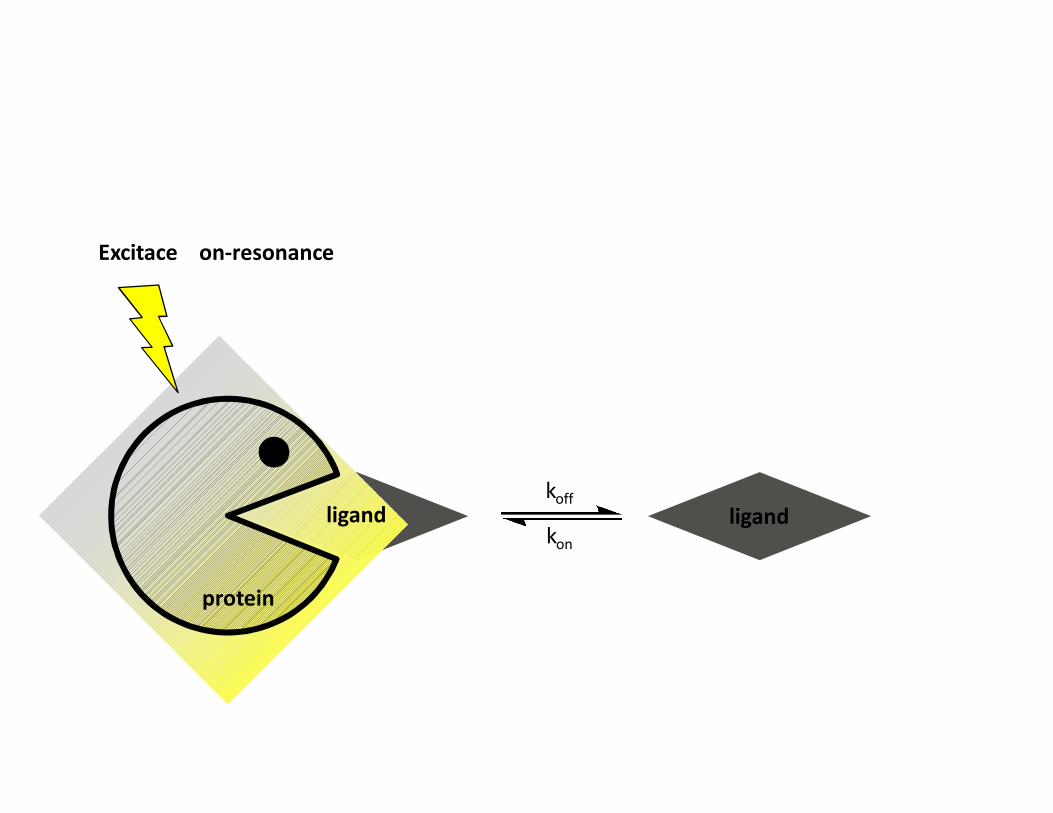
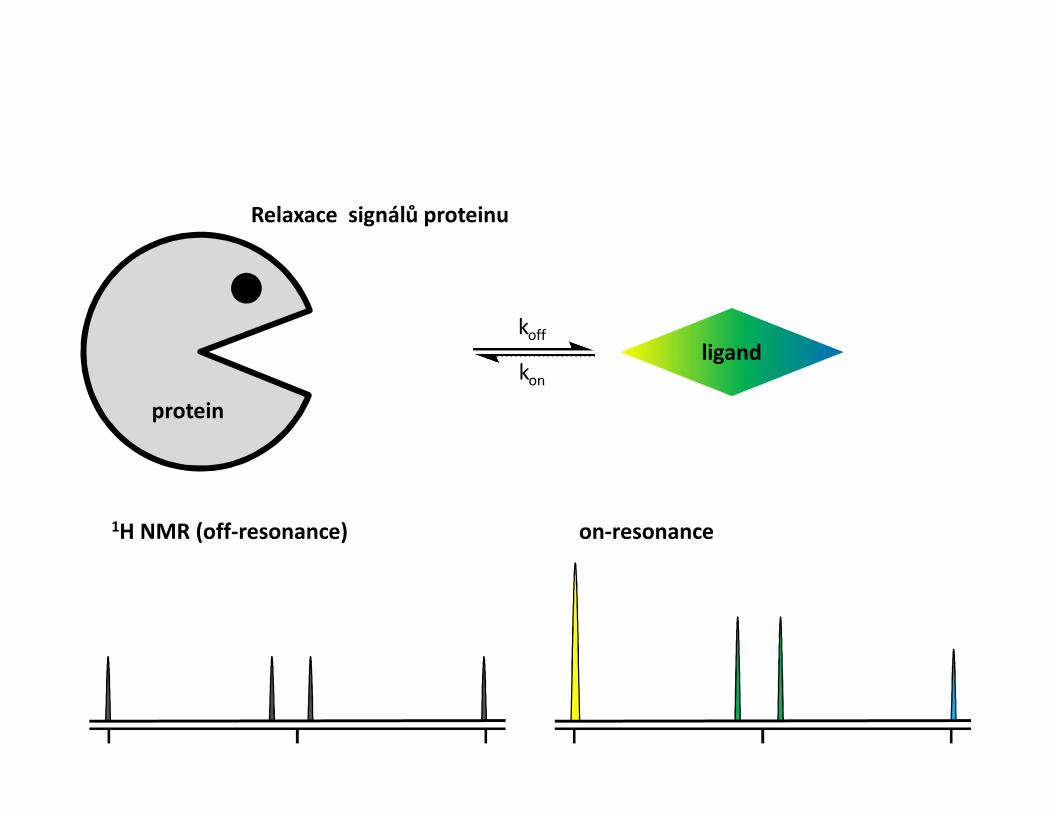
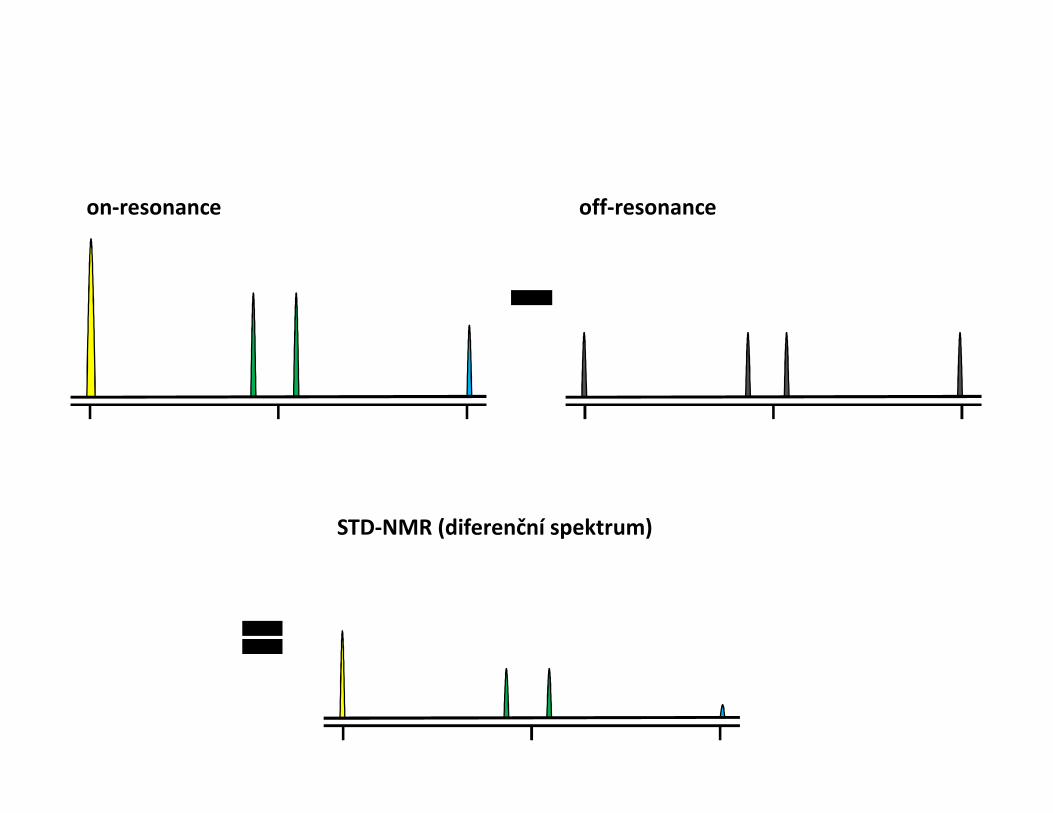
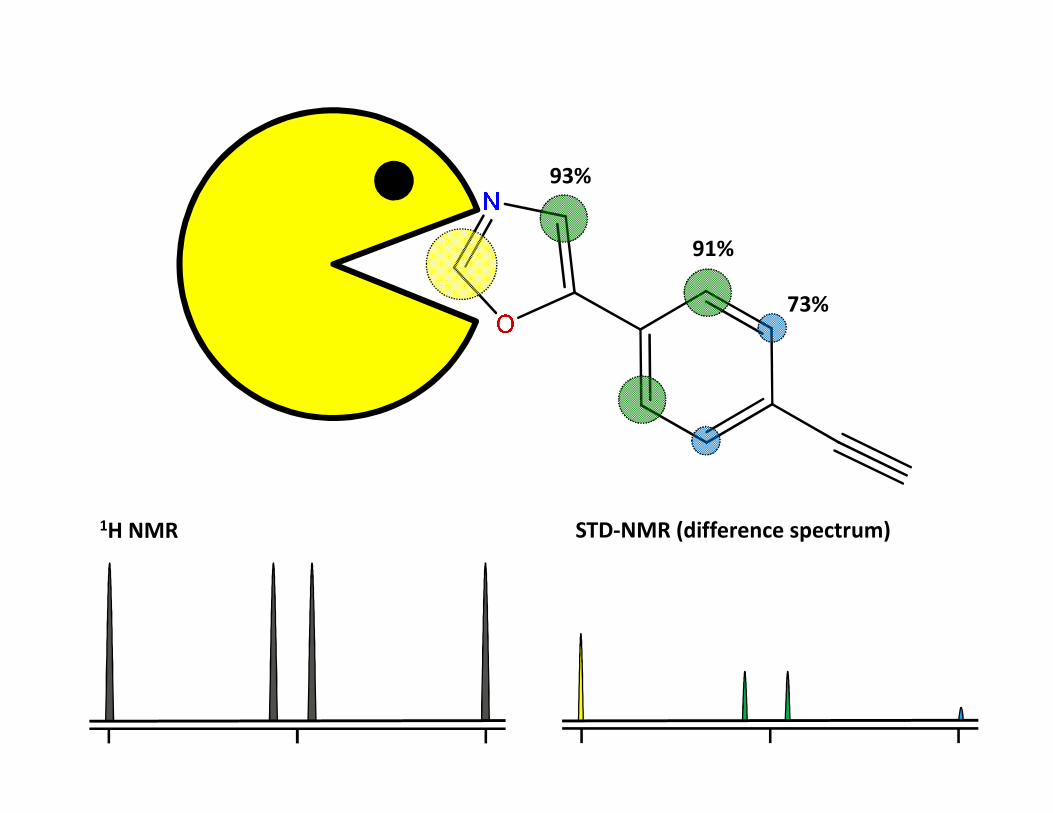
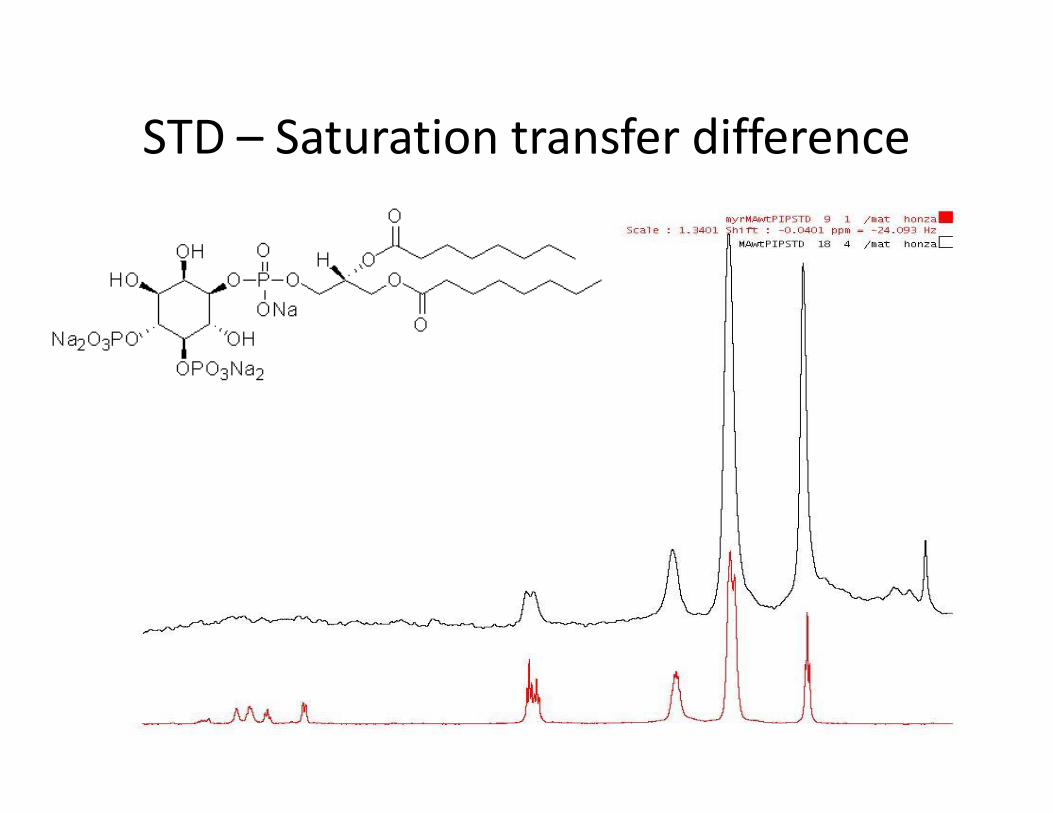
STD – Saturation transfer difference• Interakce protein-malá molekula• Ozáření proteinu a přenos magnetizace na ligand během interakce• Přebytek ligandu• Zjistíme, která část ligandu interaguje• Odhad KD
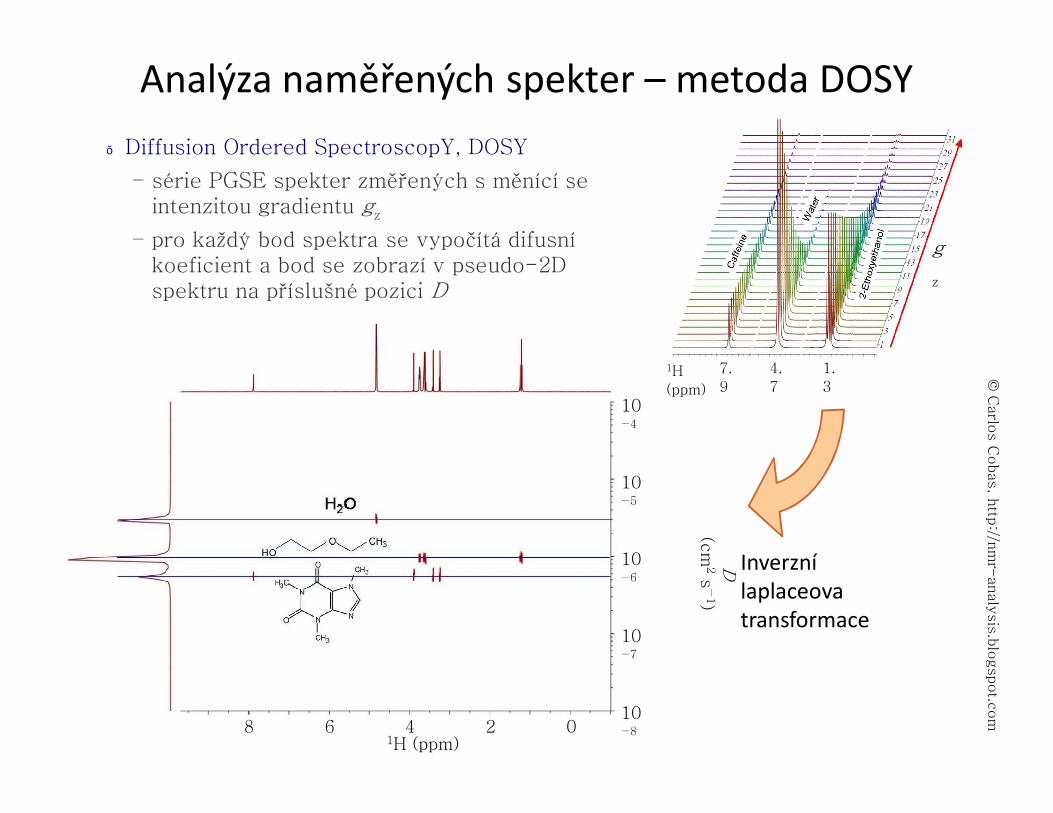
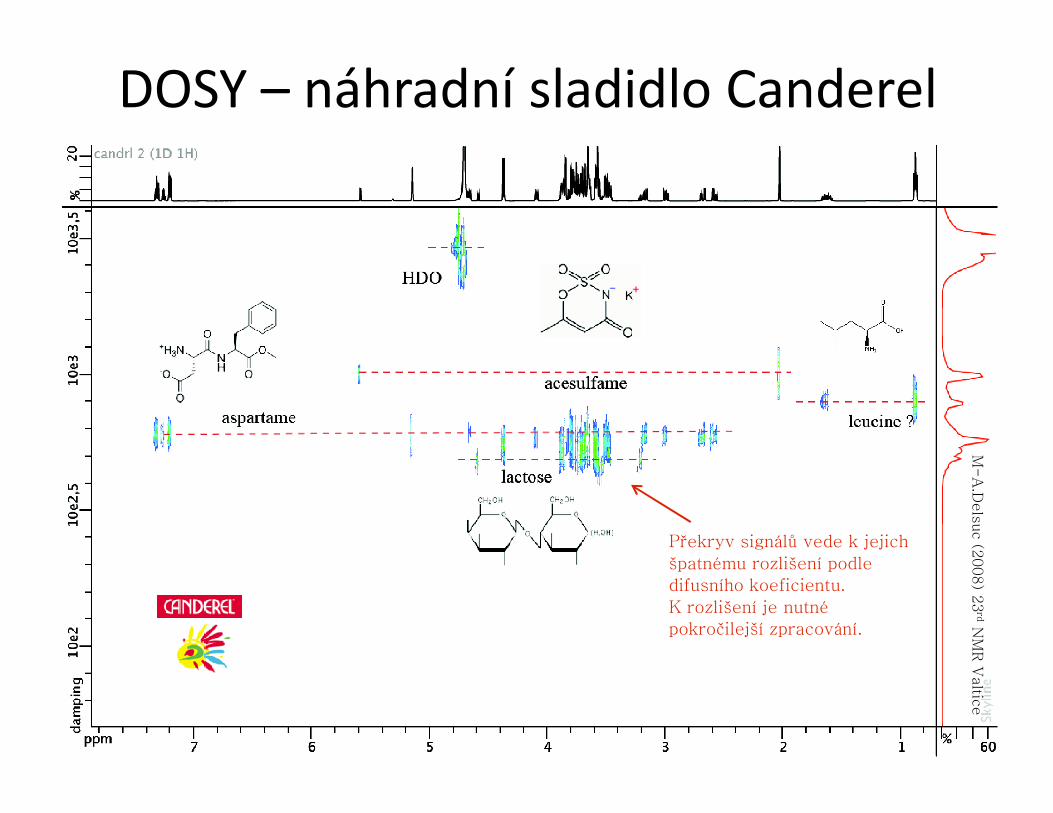
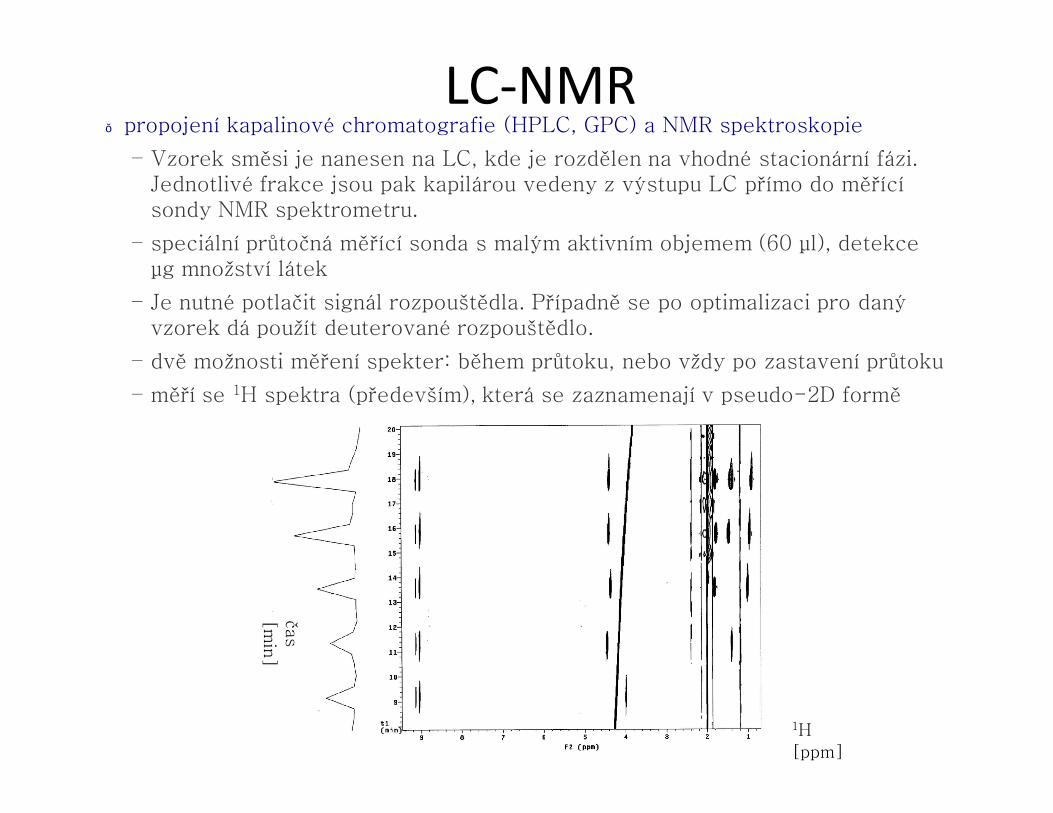
LC-NMR● propojení kapalinové chromatografie (HPLC, GPC) a NMR spektroskopie
– Vzorek směsi je nanesen na LC, kde je rozdělen na vhodné stacionární fázi. Jednotlivé frakce jsou pak kapilárou vedeny z výstupu LC přímo do měřící sondy NMR spektrometru.
– speciální průtočná měřící sonda s malým aktivním objemem (60 μl), detekce μg množství látek
– Je nutné potlačit signál rozpouštědla. Případně se po optimalizaci pro daný vzorek dá použít deuterované rozpouštědlo.
– dvě možnosti měření spekter: během průtoku, nebo vždy po zastavení průtoku
– měří se 1H spektra (především), která se zaznamenají v pseudo-2D formě
1H
[ppm]ča
s
[min
]
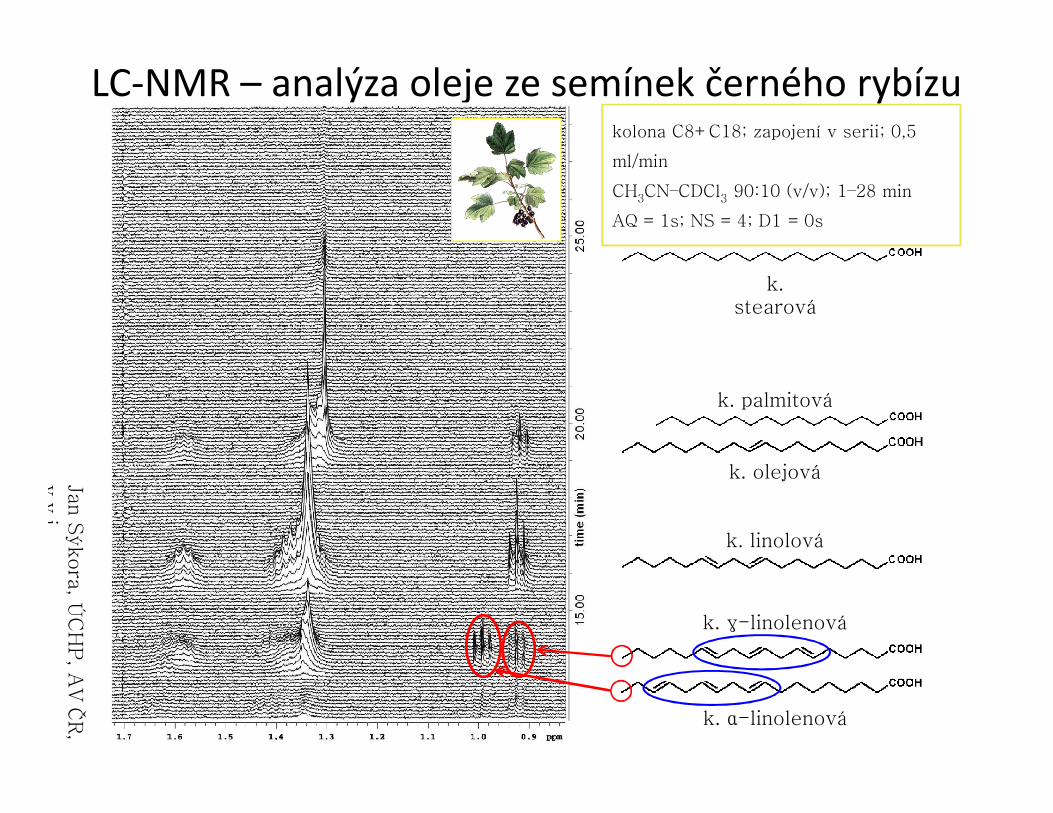
LC-NMR – analýza oleje ze semínek černého rybízu
k. stearová
k. palmitová
k. olejová
k. linolová
k. γ-linolenová
k. α-linolenová
kolona C8+C18; zapojení v serii; 0,5
ml/min
CH3CN–CDCl3 90:10 (v/v); 1–28 min
AQ = 1s; NS = 4; D1 = 0s
Jan S
ýkora
, ÚC
HP, A
V Č
R,
v.v
.i.
Průmyslové aplikace – obsah vody/oleje – NMR relaxometrie● Využívá výrazně odlišné doby
relaxace/rychlosti difuze dvou látek ve směsi
● Není potřeba vysoké rozlišení – stačí levný spektrometr s trvalým magnetem
● Použitelné na veškeré potraviny s vlhkostí pod 15%
● Odečítá se z kalibrační křivky (uložená v paměti přístroje, pro různé systémy různá)
● emulze „olej ve vodě“
– Relaxační čas T2 volného oleje je mnohem kratší než T2 volné vody. Metoda mnohonásobného spinového echa (CPMG),
– Po odečtení převažující dlouhorelaxujícíkomponenty (H2O) se získá obsah oleje extrapolací křivky (b) do t = 0. Pro získání hmotnostního zlomku je ještě nutný kalibrační faktor.
Relaxometr fi. Bruker
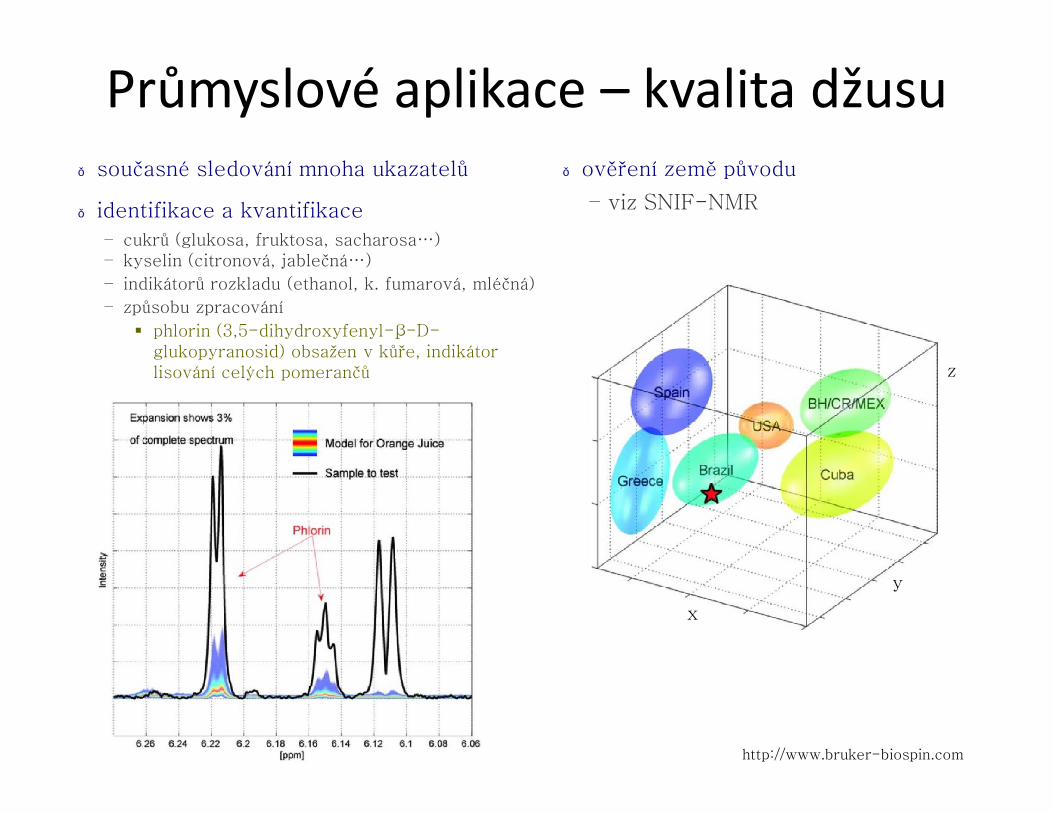
Průmyslové aplikace – kvalita džusu● současné sledování mnoha ukazatelů
– indikátorů rozkladu (ethanol, k. fumarová, mléčná)
– způsobu zpracování
§ phlorin (3,5-dihydroxyfenyl-β-D-glukopyranosid) obsažen v kůře, indikátor lisování celých pomerančů
● ověření země původu
– viz SNIF-NMR
http://www.bruker-biospin.com
x
y
z
NMR spektroskopie jako nástroj forenzní analýzy
● Použití NMR spektroskopie ve forenzní analýze je limitováno nízkou citlivostí metody a finanční náročností nákupu a provozu NMR spektrometru (národní referenční laboratoře)
● Hlavní využití:
– Průkaz zakázané látky
§ Návykové látky
§ Často je nutné prokázat, že se jedná přesně o zakázanou látku (MS/IČ nestačí)
§ Problém „legálních drog“ (látky účinné, ale dosud nezakázané)
– Průkaz falšování potravin
§ Ověření původu
§ Přítomnost zakázané/jiné než deklarované látky
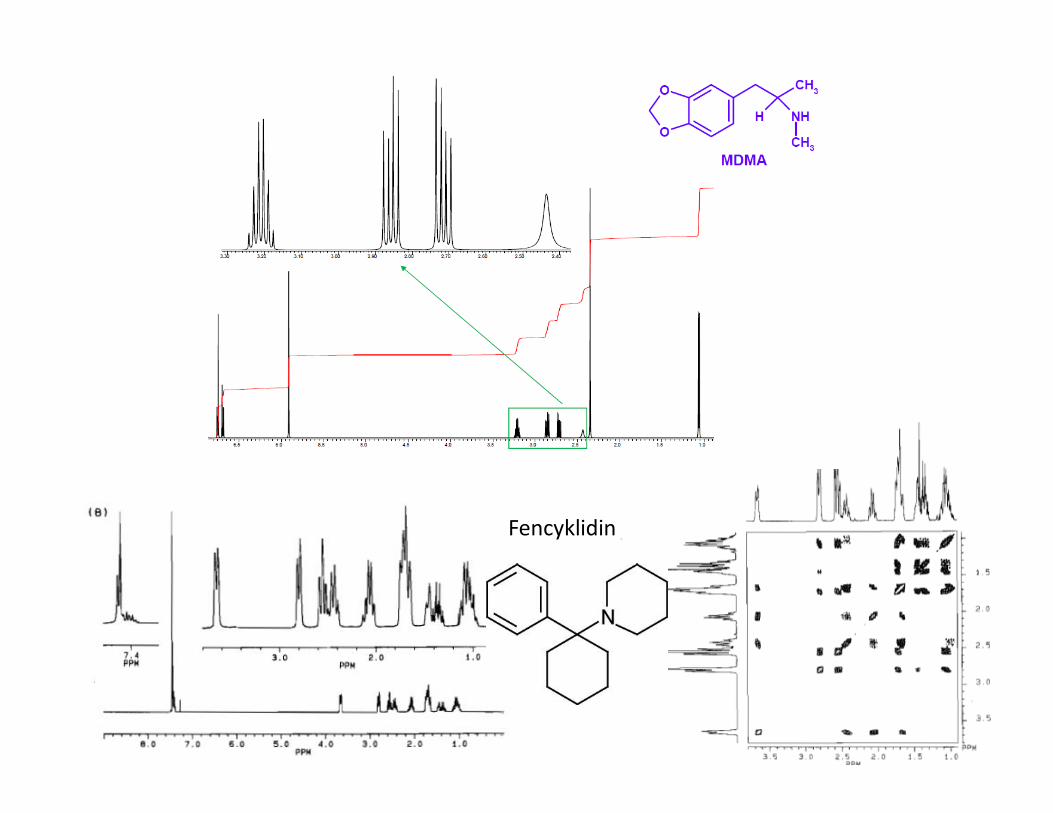
Průkaz zakázaných látek● Použití NMR spektroskopie ve forenzní analýze je limitováno nízkou citlivostí
metody a finanční náročností nákupu a provozu NMR spektrometru (národní referenční laboratoře)
● Prokázání přítomnosti látky
● Určení čistoty/nečistot, optické čistoty, zda se jedná o sůl
§Použití při vyšetřování původu látky
● Klasické experimenty 1H, 13C, vícerozměrné experimenty, posunová činidla
● Knihovny spekter známých látek
Fencyklidin
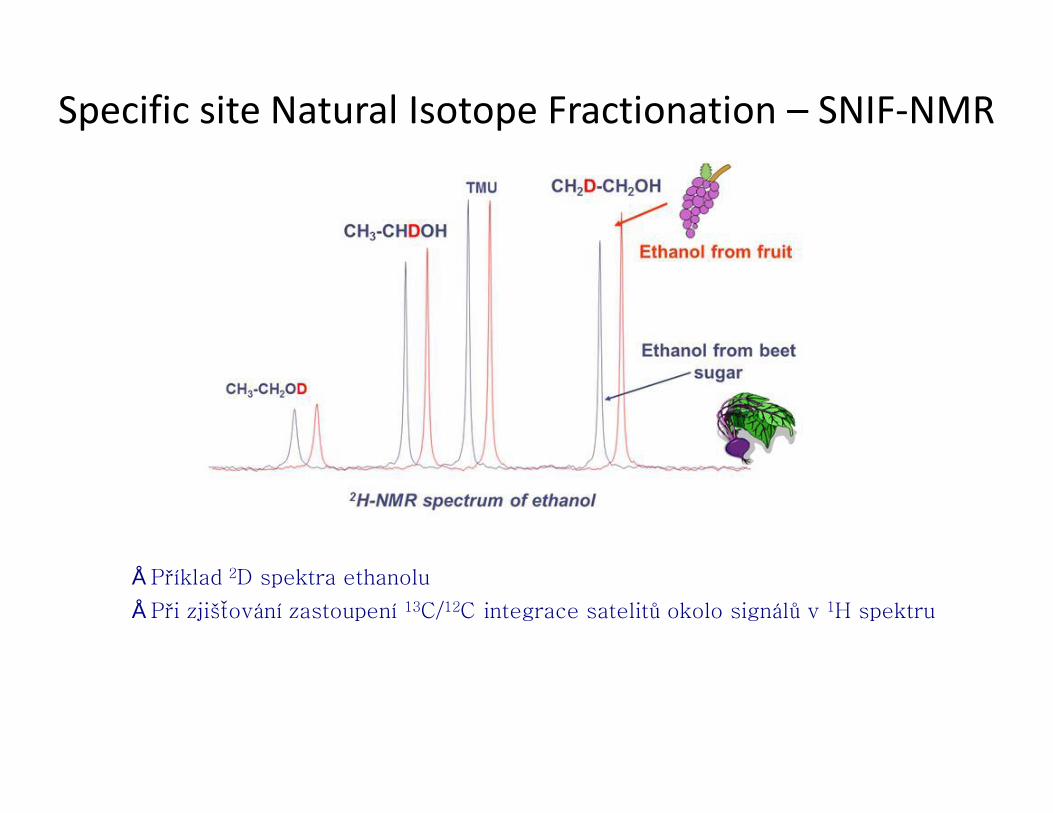
Specific site Natural Isotope Fractionation – SNIF-NMR
● sledování rozdílného izotopického zastoupení na jednotlivých místech molekuly
● Izotopické rozdělení závisí na původu příslušné molekuly. Např. D/H vody v Nantes (Francie) je 150 ppm, v Antarktidě 89 ppm. Je rozdíl např. v odpařování H2O a HDO.
● SNIF-NMR: určení poměru D/H
● odchylka isotopů (isotope deviation), δ
● alternativní metoda: hmotnostní spektrometrie (v praxi se tak měří zastoupení 13C/12C)
● použití: ověření pravosti a původu potravin
V.SMOW = Vienna
Standard Mean OceanWater
Materiály a obrázky laskavě poskytli J. Mazáč a P. Havelec z Celní laboratoře MF ČR.
Distribuce isotopů 13C a 2H v přírodě
Specific site Natural Isotope Fractionation – SNIF-NMR● Metoda schválena pro (H/D):
– Ovocné šťávy
§ Obvykle před měřením jsou cukry fermentačně převedeny na ethanol
§ Jediná metoda pro průkaz cukru z C3 nebo C4 rostlin
– Víno
– Vanilin
– Kyselina octová
• V kombinaci s 13C/12C je možné odlišit i cukry z C4 od CAM rostlin (falšování tequilly)
• Měření 2D spektra a integrace jednotlivých píků vůči standardu
• Při zjišťování zastoupení 13C/12C integrace satelitů okolo signálů v 1H spektru
• Alternativa – MS spektroskopie (hlavně 13C/12C), výhodnou NMR je zjištění poměrů v jednotlivých skupinách atomů - přesnější
Specific site Natural Isotope Fractionation – SNIF-NMR
• Příklad 2D spektra ethanolu
• Při zjišťování zastoupení 13C/12C integrace satelitů okolo signálů v 1H spektru