Université Pierre & Marie Curie LICENCE DE SCIENCES ET TECHNOLOGIE Mention CHIMIE LC 304 CHIMIE INORGANIQUE 2 DOCUMENT DE COURS Chimie de coordination Enseignants Pierre GOUZERH Souhir BOUJDAY, Xavier CARRIER, Sophie CASSAIGNON, Corinne CHANEAC, Anne DAVIDSON, Ammy El FAKIR, Jérôme LONG, Lionel NICOL, Lorenzo STIEVANO Année universitaire 2007-2008
Transcript
Université Pierre & Marie Curie
LICENCE DE SCIENCES ET TECHNOLOGIE
Mention CHIMIE
LC 304 CHIMIE INORGANIQUE 2
DOCUMENT DE COURS
Chimie de coordination
Enseignants
Pierre GOUZERH Souhir BOUJDAY, Xavier CARRIER, Sophie CASSAIGNON, Corinne CHANEAC, Anne DAVIDSON, Ammy El FAKIR, Jérôme LONG, Lionel NICOL, Lorenzo STIEVANO
Année universitaire 2007-2008
2
LC 304 - Cours de Chimie de Coordination (10h) 2007-2008
Introduction Chapitre I Isoméries I-1 Métaux, ligands et complexes I-2 Coordinences et géométries usuelles I-3 Isoméries I-3-a Isoméries de distribution des ligands I-3-b Isoméries de liaisons I-3-c Stéréoisoméries Chapitre II Interaction métal-ligand II-1 Modèle ionique : modèle du champ cristallin II-1-a Levée de dégénérescence des orbitales d II-1-a-a Complexes octaédriques et tétraédriques II-1-a-b Extension à d'autres géométries II-1-b Energie de stabilisation due au champ cristallin (ESCC) II-1-c Préférences stéréochimiques II-2 Modèle covalent : modèle des orbitales moléculaires (appliqué à une fonction M-L) II-2-a Exemple de ligand exclusivement donneur s (NH3) II-2-b Exemple de ligand donneur σ et donneur π (O2-) II-2-c Exemple de ligands donneurs σ et accepteurs π (CO, N2, CN-) II-2-d Diagrammes d’OM des complexes [ML6] II-3 Série spectrochimique Chapitre III Stabilité des complexes de coordination III-1 Facteurs enthalpiques et entropiques III-2 Facteur statistique III-3 Effet chélate, effet macrocyclique, effet cryptate
III-4 Influence du cation métallique : rôle de l’ESCC. Série d’Irving-Williams III-5 Théorie HSAB (Hard and Soft Acids and Bases) III-6 Applications Chapitre IV Réactions de substitution IV-1 Classification des mécanismes : D, A, I. Critères distinctifs IV-2 Complexes tétraédriques IV-3 Complexes plan-carrés : type A IV-4 Complexes octaédriques IV-4-a Echange du solvant. Rôle de l'ESCC : énergie d'activation due au champ cristallin (EACC) IV-4- Hydrolyse des complexes de cobalt(III) : type D Chapitre V Réactions de transfert d'électron V-1 Réactions de type sphère externe - Relation de Marcus V-2 Réactions de type sphère interne Chapitre VI Synthèse des complexes de coordination VI-1 Réactions de substitution et/ou de transfert d’électron(s) VI-1-a Synthèse des complexes de platine(II) : application de l’effet trans VI-1-b Exemples divers VI-1-c Catalyse par transfert d’électron VI-2 Réactivité des ligands coordonnés – Effet template – Auto-assemblage VI-2-a Activation vs. stabilisation VI-2-b Complexes macrocycliques VI-2-c Complexes macrobicycliques VI-2-d Auto-assemblage
3
4
5
Chapitre I — Isoméries I-1 Métaux, ligands et complexes I-1-a Configurations électroniques des métaux de transition d Série G 3 4 5 6 7 8 9 10 11 12 1 [Ar] Sc Ti V Cr Mn Fe Co Ni Cu Zn 3d 1 2 3 5 5 6 7 8 10 10 4s 2 2 2 1 2 2 2 2 1 2 2 [Kr Y Zr Nb Mo Tc Ru Rh Pd Ag Cd 4d 1 2 4 5 5 7 8 10 10 10 5s 2 2 1 1 2 1 1 0 1 2 3 [Xe) La Hf Ta W Re Os Ir Pt Au Hg 4f 0 14 14 14 14 14 14 14 14 14 5d 1 2 3 4 5 6 7 9 10 10 6s 2 2 2 2 2 2 2 1 1 2 La configuration électronique d’un ion de transition, simple ou complexe est de la forme dn avec n = G – x où x est le nombre d’oxydation (algébrique) du métal. I-2 Coordinences et géométries usuelles Pour les éléments de transition, les coordinences usuelles sont 2, 4 et 6, mais on rencontre aussi les coordinences 1, 3, 5, 7, 8 et 9. La coordinence ne peut excéder 9 pour un élément de transition d, mais on rencontre des coordinences plus élevées pour les lanthanides. Coordinence Polyèdre de coordination Exemples 1 Cu{2,4,6-C6H2PPh3) 2 linéaire [Cu(NH3)2]+
3 plan trigonal [Fe{N(SiMe3)2}3] 4 tétraédre [FeCl4]-, [Ni(PF3)4] 4 plan carré [Ni(CN)4]2-, [PtCl4]2-
5 bipyramide trigonale [CuCl5]3-, [CdCl5]3-
5 pyramide à base carrée [Ni(CN)5]3-, [WOCl4] 6 octaèdre [TiF6]2-
12 icosaèdre [Ce(NO3)6]3- Remarques : le concept de coordinence est parfois ambigu : • Dans le complexe [Cu(NH3)6]2+, 4 liaisons Cu-N valent 207 pm alors que les deux autres valent 262 pm ; faut-il alors parler de coordinence 6, de coordinence 4, ou de coordinence 4+2 ? • Dans les complexes [Ce(NO3)5]2- et [Ce(NO3)6]3-, l'ion nitrate est bidente, mais il n'est pas absurde de le considérer comme occupant un seul site de coordination : les deux complexes peuvent alors être respectivement décrits comme une bipyramide trigonale et un octaèdre.
6
Polyèdres de coordination I-3 Isoméries I-3-a Isomérie de distribution des ligands [PtCl2(en)2]Br2 [PtBr2(en)2]Cl2 cis-[CrCl2(H2O)4]Cl [CrCl(H2O)5]Cl2 [Cr(H2O)6]Cl3 [Co(NH3)6][Cr(CN)6] [Co(NH3)6][Cr(CN)6] [Ni(phen)3][Co(SCN)6] [Co(phen)3][Ni(SCN)6] I-3-b Isomérie de liaison [Co(ONO)(NH3)5]2+ [Co(NO2)(NH3)5]2+
[Co(NCS)(NH3)5]Cl2 [Co(SCN)(NH3)5]Cl2
[Pd(SCN)2(bipy)] [Pd(NCS)2(bipy)] I-3-c Stéréoisoméries I-3-c-α Définitions • Asymétrique : qualifie une molécule dépourvue de tout élément de symétrie. • Dissymétrique : qualifie une molécule dépourvue d'axe Sn (un axe impropre d'ordre n correspond à une ou plusieurs répétitions de la séquence suivante : rotation de 2π/n suivie d'une réflexion par rapport à un plan perpendiculaire à l'axe de rotation). • Stéréoisomères : molécules possédant le même contenu atomique, les mêmes séquences de liaisons mais différant par leur arrangement dans l'espace. • Chiralité : un composé est chiral s'il n'est pas superposable à son image dans un miroir. Un composé n'est chiral que s'il est asymétrique ou dissymétrique. • Enantiomères : un composé chiral et son image dans un miroir sont appelés énantiomères. • Diastéréo-isomères : deux stéréoisomères non énantiomères sont appelés diastéréo-isomères. Les isomères géométriques sont des diastéréo-isomères.
type diastéréo-isomères Exemples [Mabcdef] 15 [Pt(Br)(Cl)(I)(NO2)(NH3)(py)] [Ma2b2c2] 5 [Co(CN)2(NH3)2(H2O)2]+, [PtCl2(NH3)2(py)2]2+ [Ma4b2] 2 (cis et trans) [CoCl2(NH3)4]+ [Ma3b3] 2 (fac et mer) [IrCl3(PPh3)3] • Complexes plan-carrés [Ma2b2] 2 (cis et trans) [PtCl2(NH3)2] [Mabcd] 3 [Pt(NH3)(NH2OH)(py)(NO2)]+ I-3-c-γ Enantiomérie (isomérie optique) On se limitera ici aux complexes octaédriques renfermant deux ou trois ligands bidentes. Les complexes cis-[MX2(aa)2], cis-[MXY(aa)2], cis-[MX2(ab)2], cis-[MXY(ab)2], [M(aa)3], [M(ab)3]... où X et Y désignent des ligands monodentes et (aa) et (ab) des ligands bidentes, sont chiraux. La nomenclature utilise les descripteurs Δ et Λ que l'on définit de la manière suivante. [voir : Commission on the Nomenclature of Inorganic Chemistry of the I.U.P.A.C., Inorg. Chem., 1970, 9, 1-5 ; Y. Jeannin & E. Samuel, Nomenclature en chimie inorganique - Composés de coordination, Techniques de l'Ingénieur, juin 1995, Dossier K103]. Deux lignes, AA et BB, qui ne se coupent pas et qui ne sont ni parallèles ni orthogonales, définissent une hélice tracée sur un cylindre. La ligne AA est l'axe du cylindre ; la ligne BB est tangente à l'hélice. Peu importe la ligne choisie pour définir l'axe du cylindre, mais elle doit se trouver en arrière par rapport à l'autre. S'il faut tourner la ligne AA dans le sens inverse des aiguilles d'une montre pour l'amener en coïncidence avec BB (plus exactement parallèle à BB), l'hélice est notée Λ ; elle est notée Δ si la rotation s'effectue dans le sens des aiguilles d'une montre.
K. F. Purcell & J. C. Kotz, Inorganic Chemistry, Saunders, 1977, p. 641.
Pour appliquer ces considérations aux complexes octaédriques définis précédemment, on
assimile les ligands bidentes aux arêtes qu'ils sous-tendent dans l'octaèdre de coordination, et celles-ci sont repérées par des traits plus épais. On en choisit une comme ligne AA et une autre comme ligne BB.
8
K. F. Purcell & J. C. Kotz, Inorganic Chemistry, Saunders, 1977, p. 642.
A ceux qui éprouveraient des difficultés pour appliquer la procédure précédente, on peut suggérer l'approche suivante : • l'un des ligands se trouve nécessairement dans le plan de figure et il est représenté par une ligne courbe reliant les sommets correspondants. • La projection du ligand reliant le sommet supérieur au plan est représentée par un segment de droite. • La figure est alors tournée de façon que la ligne courbe se trouve en haut. Si la ligne droite descend vers la gauche, l'hélice est de type Λ, si elle descend vers la droite, elle est de type Δ.
Application de la méthode à différentes orientations des énantiomères d'un complexe de type [M(aa)3] (S. Herrero, M. A. Usón, J. Chem. Educ., 1995, 72, 1065). Remarque. Les énantiomères sont encore parfois appelés antipodes optiques, en raison de leur action sur le plan de polarisation d'une onde plane polarisée rectilignement : l'un des isomères
9
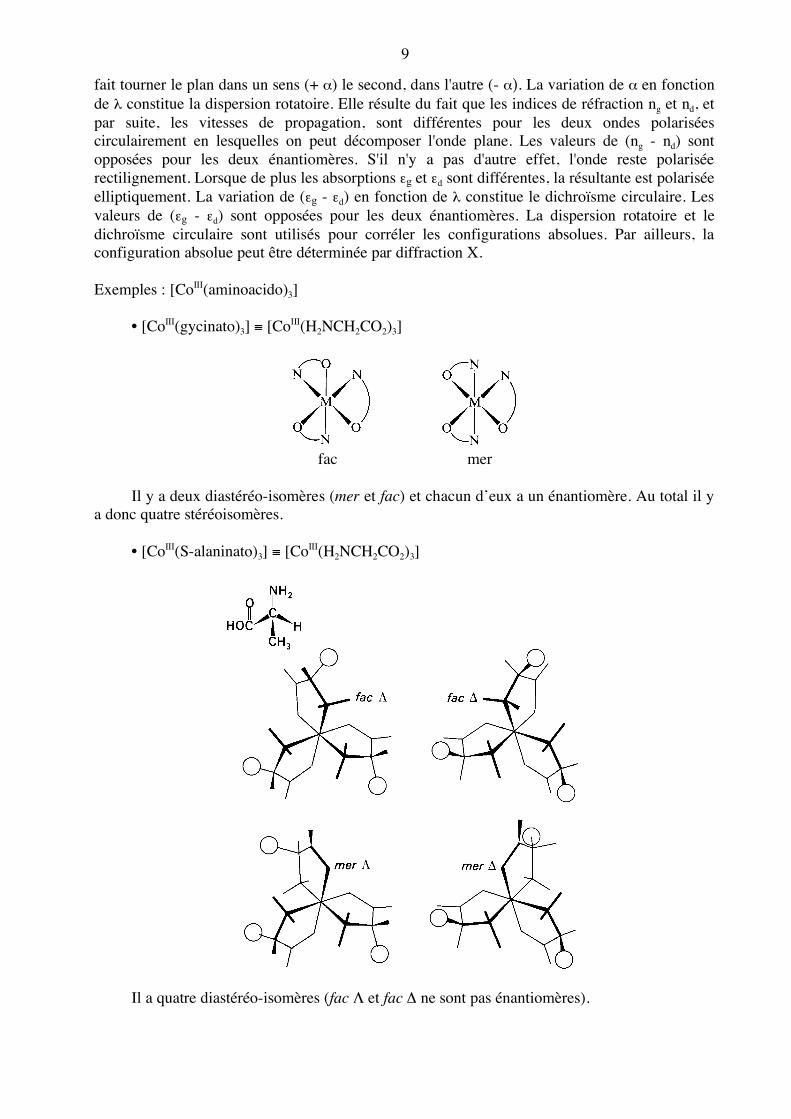
fait tourner le plan dans un sens (+ α) le second, dans l'autre (- α). La variation de α en fonction de λ constitue la dispersion rotatoire. Elle résulte du fait que les indices de réfraction ng et nd, et par suite, les vitesses de propagation, sont différentes pour les deux ondes polarisées circulairement en lesquelles on peut décomposer l'onde plane. Les valeurs de (ng - nd) sont opposées pour les deux énantiomères. S'il n'y a pas d'autre effet, l'onde reste polarisée rectilignement. Lorsque de plus les absorptions εg et εd sont différentes, la résultante est polarisée elliptiquement. La variation de (εg - εd) en fonction de λ constitue le dichroïsme circulaire. Les valeurs de (εg - εd) sont opposées pour les deux énantiomères. La dispersion rotatoire et le dichroïsme circulaire sont utilisés pour corréler les configurations absolues. Par ailleurs, la configuration absolue peut être déterminée par diffraction X. Exemples : [CoIII(aminoacido)3] • [CoIII(gycinato)3] ≡ [CoIII(H2NCH2CO2)3]
fac mer Il y a deux diastéréo-isomères (mer et fac) et chacun d’eux a un énantiomère. Au total il y a donc quatre stéréoisomères. • [CoIII(S-alaninato)3] ≡ [CoIII(H2NCH2CO2)3]
Il a quatre diastéréo-isomères (fac Λ et fac Δ ne sont pas énantiomères).
10
Spectres de dichroïsme circulaire (εg - εd) = (εl - εr) f(λ) des quatre stéréoisomères du complexe [CoIII(S-alaninato)3] I-3-c-δ Conformation des cycles de chélation Un cycle de chélation M(en) peut se présenter sous deux conformations énantiomériques selon le sens de l’hélice définie par la liaison C-C et la droite passant par les deux atomes d’azote.
Un complexe [M(en)3]z+ peut alors exister sous huit formes (quatre paires d’énantiomères). Λδδδ Δλλλ Λδδλ Δλλδ Λδλλ Δλδδ Λλλλ Δδδδ
Trois des quatre conformations possibles de Λ-[Cr(en)3]3+
Les formes Δλλλ et Λδδδ sont généralement les plus stables mais il y a des exceptions.
11
Chapitre II — Interaction métal-ligand
II-1 Modèle ionique : modèle du champ cristallin II-1-a Levée de dégénérescence des orbitales d par le champ cristallin II-1-a-α Complexes octaédriques et tétraédriques : cf. LC205 II-1-a-β Extension à d’autres géométries
Coordinence Géométrie dx2-y2 dz2 dxy dxz dyz
1 Ligand sur axe z -3,14 5,14 -3,14 0,57 0,57 2 Linéaire, ligands sur axe z -6,28 10,28 -6,28 1,14 1,14 3 Trigonale (D3h), ligands dans le
plan xy 5,46 -3,21 5,46 -3,86 -3,86
4T Tétraèdre (Td) -2,67 -2,67 1,78 1,78 1,78 4PC Plan-carré (D4h), ligands dans le
plan xy 12,28 -4,28 2,28 -5,14 -5,14
5 Bipyramide trigonale (D3h), plan de base xy
-0,82 7,07 -0,82 -2,72 -2,72
6 Octaèdre, ligands sur axes x, y, z 6,00 6,00 -4,00 -4,00 -4,00 7 Bipyramide pentagonale (D5h),
n : nombre d’électrons d ; P : énergie d’appariement des électrons Exemple :
Estab = –2,4 !o + 2P Estab = –0,4 !o
E
Complexes à spin bas
Complexes à spin élevé
!o = P
Estab = –12/5 !o + 2P
Estab = –2/5 !oE
sta
b
!o
II-1-c Préférences stéréochimiques
1-les complexes tétraédriques sont obtenus avec les ions pour lesquels l’ESCC est nulle : d0 [VO4]3-, [MO4]2- (M = Cr, Mo, W) [MO4]- (M = Mn, Tc, Re) d5 [MnX4]2- (X = Cl, Br, I), [FeX4]- (X = Cl, Br, I, NCS) d10 [Ni(CO)4], [Co(CO)4]-, [Cu(py)4]+, [Au(PMePh2)4]+
et ceux pour lesquels l’ESCC ne défavorise pas trop le tétraèdre par rapport à l’octaèdre : d7 [CoX4]2- (X = Cl, Br, I, NCS) d8 [NiX4]2- d9 [CuX4]2- dans Cs2[CuX4] (X = Cl, Br) 2-Tous les ions de la 1ère série forment des complexes octaédriques, mais les complexes de configuration d4 haut-spin, d7 bas-spin ou d9 ne peuvent être réguliers ; la déformation de l'octaèdre de coordination (effet Jahn-Teller) peut aller jusqu'au passage à la coordinence 4 plan-carrée. 3-Dans la 1ère série, les complexes bas-spin sont relativement peu nombreux, du moins si l'on se limite aux états d'oxydation intermédiaires. Ils sont cependant plus fréquents dans le cas de la configuration d6. 4-Dans les 2ème et 3ème séries, les ions de configuration dn avec n ≤ 6 forment généralement des complexes octaédriques bas-spin : d4 [MX6]2- (M = Ru, Os ; X = F, Cl, Br) d5 [MCl6]3- (M = Ru, Os), [IrX6]2- (X = F, Cl, Br) d6 [M(CN)6]5- (M = Tc, Re), [Ru(H2O)6]2+, [Ru(NH3)6]2+
13
[M(CN)6]4- (M = Ru, Os), [M(H2O)6]3+, [MCl6]3- (M = Rh, Ir) [MX6]2- (M = Pd, Pt), En revanche, les ions pour lesquels n est supérieur à 6 forment généralement des complexes plan-carrés : d7 [RhCl2{P(o-MeC6H4)3}2] d8 [RhCl(PPh3)3], [MX4]2- (M = Pd, Pt ; X = Cl, Br, I, SCN, CN) [AuX4]- (X = F, Cl, Br, I, SCN, CN, NO3), d9 [Ag(py)4]2+
5-les effets stériques peuvent être déterminants. Par exemple, l'emploi de ligands volumineux et à champ faible favorise un environnement tétraédrique par rapport à un environnement plan-carré dans le cas de Ni2+. II-2 Modèle covalent : modèle des orbitales moléculaires Lorsque les conditions sont remplies pour qu'une interaction se produise entre deux orbitales atomiques φΑ, d'énergie EA et φB, d'énergie EB, celles-ci se combinent linéairement pour donner deux nouvelles orbitales, moléculaires cette fois. La plus stable est d'énergie inférieure au plus bas niveau initial EB ; elle est liante, ce qui se caractérise par une continuité de phase entre les atomes A et B. La plus haute est d'énergie supérieure au plus haut niveau initial EA ; elle est antiliante, ce qui se caractérise par un changement de phase entre A et B. On peut encore noter que l'énergie de stabilisation de l'orbitale liante est inférieure, en valeur absolue, à l'énergie de déstabilisation de l'orbitale antiliante. Par ailleurs, l'interaction augmente quand EA - EB diminue ; elle est maximale lorsque EA = EB. Enfin, l'énergie d'interaction croît avec l'intégrale de recouvrement S = ∫∫∫∞φAφBdτ et s'annule avec elle. Or, pour que cette intégrale ne soit pas nulle, il est nécessaire que les orbitales aient les mêmes propriétés de symétrie. II-2-a Exemple de ligand exclusivement donneur σ : NH3
Diagramme d’OM de NH3
14
Diagramme d’OM de la fonction {Ni(NH3)}2+, limité aux orbitales d : l’interaction entre
l’orbitale dz2 du métal et la HOMO de NH3 (2a1) crée deux orbitales σ et σ*
II-2-b Exemple de ligand donneur σ et donneur π : ligand oxo (O2-) On se limite à la description d'un fragment MO(x-2)+ ; l'axe z est pris suivant la liaison M-O ; la composante σ (donation) met en jeu l'orbitale pz du ligand O2- et les orbitales s, pz et dz2, supposées vacantes, du cation Mx+. La composante π (donation) met en jeu les orbitales px et py (dégénérées) du ligand O2- et les orbitales dxz et dyz, supposées vacantes, du du cation Mx+. Selon l'amplitude de la donation, l'ordre formel de la liaison peut varier entre 1 (interaction π négligeable : le ligand oxo apporte deux électrons au métal) et 3 (interaction π maximale : le ligand apporte 6 électrons au métal).
Diagramme d’OM de la fonction {CrO}4+, limité aux orbitales d
Remarques :
15
(i) Ces deux situations limites, ainsi que la situation intermédiaire où le ligand oxo apporte 4 électrons au métal) peuvent être schématisées en notation de Lewis. Dans la théorie du lien de valence, la fonction d'onde résulte de la superposition de ces trois formes. (ii) Seuls les cations de configuration d0, d1 et d2 forment des complexes oxo. Pour ces hauts degrés d'oxydation, les orbitales (n-1)d sont fortement contractées, de sorte qu'elles ne participent que faiblement aux liaisons métal-ligand ; l'interaction π met alors essentiellement en jeu les orbitales np du métal. II-2-c Exemples de ligands donneurs σ et accepteur π : CO, N2, CN- II-2-c-α Comparaison CO / N2
Les trois ligands sont isoélectroniques. La HOMO de CO est faiblement antiliante ; elle correspond essentiellement au doublet libre de l'atome de carbone. La LUMO de CO est antiliante et largement localisée sur l'atome de carbone.
Diagramme d’OM de CO
La liaison M-CO comporte deux composantes : • interaction σ par recouvrement entre la HOMO de CO (3σ) et une orbitale du métal vacante et de symétrie appropriée (par exemple dz2, pz, s, ou toute combinaison de ces orbitales, la liaison M-CO étant choisie comme axe z). Il y a transfert électronique (donation) du ligand (base de Lewis) vers le métal (acide de Lewis) ; • interaction π par recouvrement entre la LUMO de CO (2πx,2πy) et les orbitales du métal occupées et de symétrie appropriée (dxz et dyz, compte tenu du choix de z). Il y a transfert électronique (rétrodonation) du métal (celui-ci joue ici le rôle de base de Lewis) vers le ligand (acide de Lewis). Les deux composantes se renforcent mutuellement : en effet, la donation augmente à la fois le caractère basique du métal et le caractère acide du ligand. La rétrodonation se traduit par le renforcement de la liaison M-C et l'affaiblissement de la liaison CO. La fréquence de la vibration d'élongation CO renseigne sur le mode de coordination du ligand CO et sur l'amplitude de la rétrodonation : plus la rétrodonation est importante, plus la fréquence de la vibration d’élongation du ligand CO est abaissée.
16
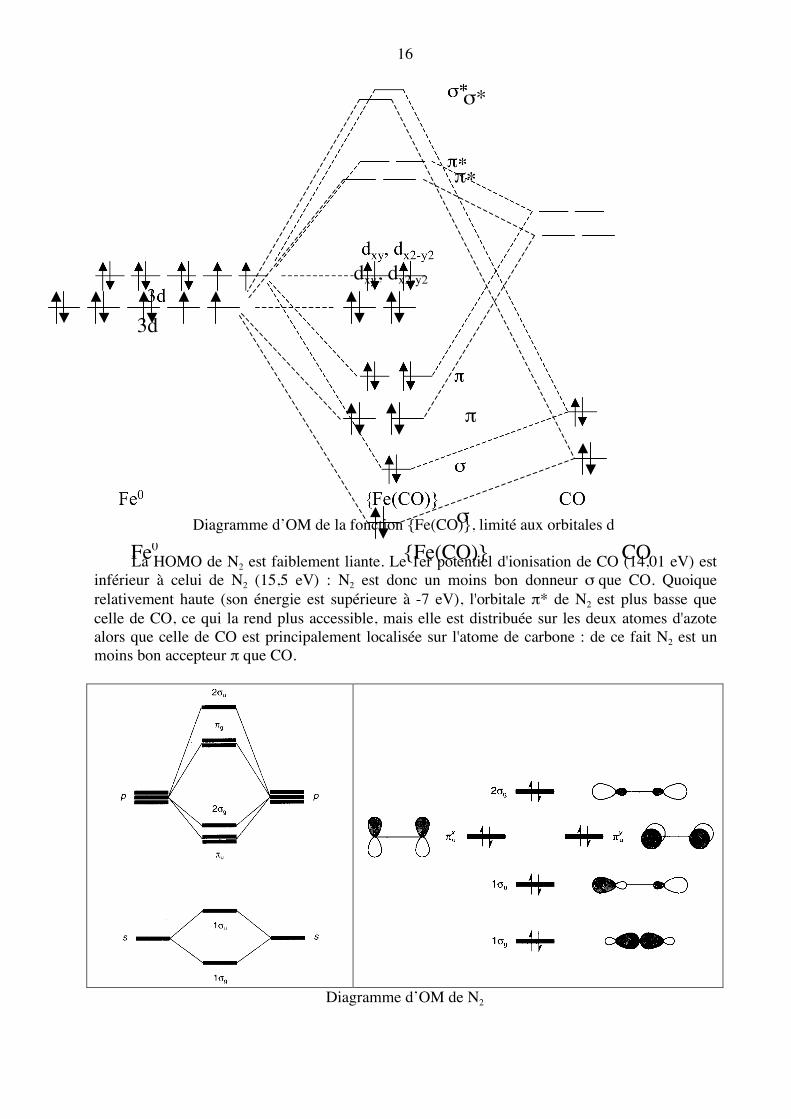
Diagramme d’OM de la fonction {Fe(CO)}, limité aux orbitales d
La HOMO de N2 est faiblement liante. Le 1er potentiel d'ionisation de CO (14,01 eV) est inférieur à celui de N2 (15,5 eV) : N2 est donc un moins bon donneur σ que CO. Quoique relativement haute (son énergie est supérieure à -7 eV), l'orbitale π* de N2 est plus basse que celle de CO, ce qui la rend plus accessible, mais elle est distribuée sur les deux atomes d'azote alors que celle de CO est principalement localisée sur l'atome de carbone : de ce fait N2 est un moins bon accepteur π que CO.
Diagramme d’OM de N2
dxy, dx2-y2
3d
σ
π
π∗
σ*
Fe0 {Fe(CO)} CO
17
Les données structurales (M-C vs M-N, CO vs NN) et spectroscopiques (νCO vs νNN) confirment que N2 est à la fois un moins bon donneur σ et un moins bon accepteur π que CO. On comprend ainsi que les complexes du diazote sont beaucoup moins stables que les métaux carbonyle : par exemple, [Ni(CO)4] est obtenu par réaction de CO sur le nickel à température ordinaire alors que [Ni(N2)4] n'a été mis en évidence qu'en matrice d'argon à très basse température. II-2-c-γ Comparaison CO/N2/CN-
Le caractère donneur σ diminue dans l'ordre : CN- > CO > N2
Le caractère accepteur π diminue dans l'ordre : CO > N2 > CN-. II-2-d Diagrammes d’OM des complexes [ML6] (cf Introduction à la Chimie du Solide)
Ligands donneurs σ
Ligands donneurs σ et π
18
Ligands donneurs σ et accepteurs π
II-3 Retour sur la série spectrochimique Δo est d'autant plus grand que les orbitales eg sont déstabilisées et que les orbitales t2g sont stabilisées. On comprend ainsi pourquoi les ligands à champ fort sont, soit des ligands fortement donneurs σ (H-, CH3
-), soit des ligands accepteurs π (o-phen, CO...). A l'inverse, les ligands donneurs π (Cl-, I-...) déstabilisent les orbitales t2g et sont donc des ligands à champ faible.
19
Chapitre III — Stabilité des complexes de coordination
La formation d'une liaison de coordination est une réaction acide-base au sens de Lewis. En règle générale, un accroissement de la basicité du ligand et/ou de l'acidité du métal entraîne un accroissement de la stabilité du complexe. Remarque : la stabilité est une notion thermodynamique qui ne doit pas être confondue avec l’inertie, notion cinétique (cf. chapitre IV). III-1 Facteurs enthalpiques et entropiques III-1-a Constantes de formation et diagrammes de distribution • Constantes successives et globales
La stabilité d'un complexe est généralement exprimée par sa constante de formation globale βn, laquelle peut être exprimée en fonction des constantes successives Kj. MLj-1 + L MLj Kj = [MLj]/[MLj-1][L]
M + jL MLj βj = [MLj]/[M][L]j
ΔG0 = ΔH0 - TΔS0 = - RTLnβj • Diagrammes de distribution : αj = [MLj]/Σ[MLj] = βj[L]i/Σβj[L]j = f[{L]libre} • Nombre moyen de ligands fixés : ñ = ([L]total] - [L]libre)/[M]total = ∑jβj[L]j/Σβj[L]j
III-1-b Stabilisation enthalpique
On considère la réaction : [M(H2O)6]2+ + NH3 [M(H2O)5(NH3)]2+ + H2O M ΔH0, kJ mol-1 ΔS0, J K-1 mol-1 ΔG0, kJ mol-1
NH3 remplace H2O car la liaison M-NH3 est plus stable que la liaison M-OH2. III-1-c Stabilisation entropique On considère la réaction : [Fe(H2O)6]3+ + X- [MX(H2O)5]2+ + H2O
Le terme enthalpique est défavorable ; en revanche, le terme entropique est favorable : les ions halogénures (surtout l'ion fluorure) sont fortement solvatés ; leur effet "structurant" disparaît avec leur complexation, ce qui entraîne une augmentation de l'entropie du solvant. III-2 Facteur statistique En règle générale, Kj > Kj+1. Si ΔHj et ΔSj peuvent être considérés comme indépendants de j, la décroissance observée peut s'expliquer à partir de considérations statistiques : la constante Kj est proportionnelle au nombre de molécules d'eau restant à remplacer dans le complexe [MLj-1] et inversement proportionnelle au nombre de ligands L susceptibles de se dissocier dans le complexe [MLj].
• Pour un ligand monodente et une coordinence N, on en déduit :
• Pour un complexe octaédrique et un ligand bidente, on attend K1/K2 = 4,8 et K2/K3 = 9,4.
Toute anomalie manifeste dans la variation des constantes de stabilité successives des complexes formés par un cation métallique et un ligand donnés traduit une variation de la coordinence ou de l'état de spin du complexe. Exemple : [CdBrj-1(H2O)N+1-j](3-j)+ + Br- [CdBrj(H2O)N-j](2-j)+ + H2O K1 = 1,56 K2 = 0,54 K3 = 0,06 K4 = 0,37 K1 > K2 > K3 < K4. L'anomalie s'explique par un changement de la coordinence : les complexes 1/1, 2/1 et 3/1 sont octaédriques alors que le complexe 4/1 est tétraédrique. [CdBr3(H2O)3]- + Br- [CdBr4]2- + 3H2O Le terme entropique de cette réaction est favorable ; il en résulte un accroissement de K. III-3 Effet chélate, effet macrocyclique, effet cryptate III-3-a Effet chélate On compare les réactions : [Ni(H2O)6]2+ + 2NH3(aq) [Ni(H2O)4(NH3)2]2+ + 2H2O K1K2 = 1,1 105 ΔG0 = -27,6 kJ mol-1 [Ni(H2O)6]2+ + en(aq) [Ni(H2O)4(en)]2+ + 2H2O K1 = 4,5 107 ΔG0 = -43,6 kJ mol-1
[Ni(H2O)4(NH3)2]2+ + 2NH3(aq) [Ni(H2O)2(NH3)4]2+ + 2H2O K3K4 = 1,1 103 ΔG0 = -17,1 kJ mol-1 [Ni(H2O)4(en)]2+ + en(aq) [Ni(H2O)2(en)2]2+ + 2H2O K2 = 2,2 106 ΔG0 = -36,2 kJ mol-1 En première approximation, l’énergie de la liaison Ni-NH3 est constante. La stabilisation additionnelle est de nature entropique. Les cycles pentagonaux sont les plus stables. Les cycles hexagonaux sont eux aussi raisonnablement stables et sont même favorisés lorsqu'il peut y avoir délocalisation électronique (exemple du ligand acétylacétonate). III-3-b Effet macrocyclique On compare le pentaglyme CH3O(CH2CH2O)5CH3 (18P6) à l’éther 18-couronne-6 (18C6) : [K(18P6)]+ + 18C6 [K(18C6)]+ + 18P6 ∆G° = -23 kJ mol-1 ∆H = -2,50 J K-1 mol-1 T∆S = 20,5 kJ mol-1
Comme dans cet exemple, l’effet macrocyclique est le plus souvent un effet entropique : le travail de réorganisation conformationnel est moins important pour le ligand cyclique.
21
18-P-6 18-C-6 • Sélectivité
O O
OO
O O
O
O
O O
O
O
OO
O
O
O O
O
OO
O
12-couronne-415-couronne-5
18-couronne-6
21-couronne-7
18-cown-6 (cavity 1.38 Å)
log
K d
ans
MeO
H
III-3-c Effet cryptate (macrobicyclique) On compare le ligand macrobicyclique [2,2,2] et le ligand macrocyclique résultant de l'ouverture de l'un des ponts (voir ci-dessous). En milieu méthanol/eau (95/5) la constante de formation du complexe [K+⊂2,2,2] est de 5 ordres de grandeur supérieure à celle du complexe macrocyclique correspondant.
O O
N
O
N
O
OO
X = CH2CH2OCH2CH2OCH3 [2,2,2]
III-4 Influence du cation métallique : rôle de l'ESCC En général les constantes de stabilité : • diminuent lorsque Z augmente pour les métaux électropositifs ;
O
O
O
OO
O
22
• augmentent lorsque Z augmente pour les métaux nobles. III-4-a complexes de configuration d0
La stabilité des ions de complexes de configuration d0 peut être interprétée à l'aide d'un modèle électrostatique (interactions ion-ion et ion-dipôle). Dans ce modèle, la stabilité augmente lorsque la charge des ions augmente et lorsque leur rayon diminue. Les complexes sont d'autant plus stables que : • l'état d'oxydation du métal est plus élevé : Ti4+ > Sc3+ > Ca2+ > K+
• que le rayon du cation est plus petit : Li+ > Na+ > K+ Mg2+ > Ca2+ > Sr2+
Al3+ > Sc3+ > Y3+
III-4-b Stabilité des ions complexes à couche d incomplète : rôle de l'ESCC En l'absence de levée de dégénérescence des orbitales d, on devrait observer une augmentation monotone de K lorsqu'on parcourt les séries de gauche à droite : en effet, Zeff augmente et r diminue. En réalité, si l'on porte log K en fonction de Z, et si l'on trace la courbe monotone passant par les points représentatifs des ions d0, d5 et d10, on constate que tous les autres points se situent au-dessus.
Exemple : [M(H2O)6]2+ + j H2NCH2CH2NH2 [M(en)j(H2O)6-2j]2+ + 2jH2O
Si l'on se limite à la deuxième partie de la série, l'ordre de stabilité est en général le suivant : Mn2+ < Fe2+ < Co2+ < Ni2+ < Cu2+ > Zn2+
Cette série (série d'Irving-Williams) reflète principalement les variations de ∆H0. L'ordre est le même pour des ligands azotés et pour les ligands oxygénés. On observe des exceptions pour les
23
ligands qui forment des complexes de spin faible avec certains cations et pour les ligands qui imposent une contrainte géométrique ou stérique. Pour les ligands du type di-imine (bpy, terpy, phen...), l'ordre habituel de stabilité est perturbé par la variation de l'état de spin des complexes de Fe(II). L'accroissement de stabilité de la configuration t2g
6 résulte en partie de l'augmentation de l'interaction σ et en partie de la rétro-coordination (interaction π).
III-5 Théorie HSAB (Hard and Soft Acids and Bases) Références
• S. Ahrland, J. Chatt, N.R. Davies, Chem. Soc. Rev., 12 (1958) 265. • R. G. Pearson, J. Chem. Ed., 45 (1968) 581 & 643. R. G. Pearson, J. Chem. Ed., 64 (1987) 561. R.G. Pearson, Inorg. Chem., 27 (1988) 734. • R.G. Parr & R.G. Pearson, J. Am. Chem. Soc., 105 (1983) 7512. Deux approches ont été proposées, qui aboutissent à des classifications similaires. • Selon Ahrland, Chatt & Davies, les cations peuvent être rangés dans deux classes selon qu'ils forment préférentiellement des complexes avec les donneurs de la seconde période (classe a) ou avec ceux de la troisième période (classe b).
Classe a Classe b F > Cl > Br > I O >> S > Se > Te N >> P > As > Sb
F << Cl < Br < I O << S ~Se ~Te N << P > As > Sb
• La théorie des acides et des bases durs et mous (théorie HSAB, Pearson) distingue : - Les acides durs dont le comportement est parallèle à celui du proton. Ils se rencontrent parmi les cations de pré-transition ou les cations de transition de haut degré d'oxydation. Leurs électrons sont peu polarisables. Ils manifestent une préférence pour les bases dures (ligands donneurs σ et donneurs π). On observe une corrélation entre K et la fonction ξ = z2/r : les interactions sont plutôt de nature électrostatique ; elles peuvent toutefois devenir covalentes lorsque le pouvoir polarisant du cation est très élevé.
- Les acides mous qui se trouvent parmi les cations de transition "lourds" ou de bas degré d'oxydation et les cations du bloc p (configurations d6, d8, d10, d10s2). Leurs électrons sont polarisables. Les cations mous manifestent une préférence pour les bases molles (ligands accepteurs π). On observe une corrélation entre K et la polarisabilité du ligand : les interactions sont plutôt de nature covalente.
24
• Les complexes les plus stables sont formés entre les acides durs et les bases dures (interaction électrostatique) et entre les acides mous et les bases molles (interaction covalente). • Une interaction mou / dur produit des complexes peu stables. • Effet symbiotique : un cation relativement dur (ou intermédiaire) est rendu plus dur interaction avec une base dure et plus mou par interaction avec une base molle.
• Relation entre dureté et électronégativité. Parr & Pearson ont proposé la définition suivante de la dureté :
! = 1
2(!µ
!N)"
µ = (
!E
!N)! = - I + A
2 = - "
où µ est le potentiel chimique électronique, E l'énergie du système, N le nombre d'électrons du système et v le potentiel créé par les noyaux. I et A désignent respectivement l'énergie d'ionisation et l'affinité électronique du système considéré. On peut en déduire :
! = 12(!µ
!N)" = 1
2(!2E
!N2)" = I - A
2 La dureté d'un système dépend donc de la différence d'énergie entre ses OMs frontières (HOMO et LUMO) ; plus celle-ci est faible, plus le système est déformable, donc mou. III-6 Applications III-6-a Prévision des réactions acido-basiques En fonction des positions relatives des OMs frontières des deux espèces antagonistes, il est possible d'envisager quatre types d'interaction : acide dur + base dure, acide mou + base molle, acide dur + base molle, acide mou + base dure. On ne retiendra que les deux premières qui correspondent à des interactions typiques, les deux autres correspondant à des effets moyens. • Réaction entre un acide dur et une base dure : l'interaction est principalement de nature coulombienne ; l'adduit est de type ionique (cependant, le caractère covalent peut devenir important lorsque la base est à la fois donneur σ et π). • Réaction entre un acide mou et une base molle : le recouvrement entre la LUMO de la base et la HOMO de l'acide est important ; l'adduit est de type covalent. III-6-b Distribution des éléments (classification de Goldschmidt) Les cations durs correspondent aux éléments lithophiles : ils sont associés à des bases dures (chlorures, oxydes, sulfates, carbonates, silicates) et se rencontrent principalement dans la croûte terrestre (lithosphère). Les cations mous correspondent aux éléments chalcophiles : ils sont associés à des bases molles (sulfures, séléniures, tellurures). III-6-c Stabilisation d'un état d'oxydation donné • Les hauts degrés d'oxydation (cations durs) sont stabilisés par les ligands durs : F-, O2-... ([MnO4]- ...) • Les bas degrés d'oxydation (cations mous) sont stabilisés par les ligands mous : CO, PR3. ([Mn(CO)5]- ...)
25
Remarque : le caractère covalent des liaisons métal-ligand est plus marqué dans les complexes de bas degré d'oxydation (interaction π métal → ligand) et dans les complexes de haut degré d'oxydation (interaction π ligand → métal) que dans les complexes de degré d'oxydation moyen. III.3.d Mode de coordination des ligands ambidentes Le concept HSAB permet aussi d'expliquer le mode de liaison des ligands ambidentes. Ainsi, le ligand thiocyanate se lie préférentiellement aux cations durs par l'atome d'azote, alors qu'il se lie aux cations mous par l'atome de soufre : [Co(NCS)(NH3)5]2+ vs. [Pd(SCN)4]2-. Dans les complexes [MCl2(DMSO)2], le ligand diméthylsulfoxyde ((CH3)2SO : DMSO) se lie à Cu(II) par l'atome d'oxygène alors qu'il se lie à Pd(II) par l'atome de soufre.
26
Chapitre IV — Réactions de substitution
Les complexes très réactifs sont dits ”labiles” tandis que les complexes peu réactifs sont dits ”inertes”. Selon Taube, un complexe labile est un complexe dont les réactions sont complètes dans le temps de mélange des réactifs (de l’ordre d’une minute, à température ordinaire pour des réactions environ 0,1 mol.l-1). Un complexe est inerte si ses réactions sont trop lentes pour qu’il soit possible de les suivre par les méthodes conventionnelles, à température ordinaire. Le test de labilité le plus fiable consiste à mesurer la vitesse d’échange des ligands dans une réaction du type : [ML6] + L [ML5(L)] + L Un complexe stable peut être labile ou inerte :
Complexe Constante de formation Vitesse d’échange [Ni(CN)4]2- 1030 Très rapide [Mn(CN)6]3- 1027 Mesurable [Fe(CN)6]4- 1037 Très lente [Fe(CN)6]3- 1044 Très lente [Hg(CN)4]2- 1042 Très rapide
IV-1 Classification des réactions
IV-1-a Mécanisme dissociatif (D) LnMX LnM + X réaction directe : constante k1 réaction inverse : constante k-1 LnM + Y → LnMY (constante k2) Approximation de l'état quasi-stationnaire :
v =
d[MLnY]
dt =
k1k2[MLnX][Y]
k-1[X] + k2[Y] Deux cas limites : k2[Y] >> k-1[X] v = kobs[LnMX] kobs = k1
k2[Y] << k-1[X] v = kobs[LnMX][Y] kobs = k1k2
k-1[X]
IV-1-b Mécanisme par interchange (I) LnMX + Y LnMX,Y Equilibre rapidement atteint ; constante K. LnMX,Y → LnMY,X Etape limitante ; constante de vitesse k. LnMY,X → LnMY + X (rapide)
v =
kK[MLnX]total[Y]
1 + K[Y] [LnMX]total = [LnMX]T = [LnMX] + [LnMX,Y] (concentration totale à l'instant t) Deux cas limites : • K[Y] >> 1 ⇒ v = kobs[LnMX]T kobs = k • K[Y] << 1 ⇒ v = kobs[LnMX]T[Y] kobs = kK
27
4-1-c Mécanisme associatif (A) LnMX + Y LnMXY Réaction directe : k1 ; réaction inverse : k-1 LnMXY → LnMY + X (k2) Approximation de l'état quasi-stationnaire :
v =
d[ML5Y]
dt =
k1k2[ML5X][Y]
k-1 + k2
= kobs[ML5X][Y]
kobs =
k1k2
k-1 + k2 4-1-d Critères diagnostiques du mécanisme Influence du ligand partant Influence du ligand entrant Influence des ligands non remplacés Effets stériques Effets électroniques Influence de la charge Paramètres d'activation ΔS‡ < 0 ΔV‡ < 0 type A ΔS‡ > 0 ΔV‡ > 0 type D IV-2 Réactions de substitution dans les complexes plan-carrés : type A [ML3X] + solvant → [ML3X(solvant)] (lent, k1)
L’intermédiaire est une bipyramide trigonale où le ligand partant, le ligand entrant, et le ligand initialement en trans du ligand partant occupent les positions équatoriales. • Influence du groupe entrant trans-[Pt(py)2Cl2] + Y → trans-[Pt(py)2ClY] + Cl-
On définit un paramètre de nucléophilie, n, par la relation :
n = log
k2
k2ref
où k2ref se rapporte à une base choisie comme standard, CH3OH par exemple. La valeur de n pour
un ligand Y donné est une mesure de sa nucléophilie par rapport à CH3OH. Dans l'exemple considéré, on observe une corrélation entre le paramètre de nucléophilie et la mollesse du ligand entrant. • Influence du groupe partant
28
[PtX(dien)]+ + py [Pt(dien)py]2+ + X– (dans l'eau) X CN– I– Cl– H2O k mol–1 l s–1 1,7 10–8 2,0 10–7 3,5 10–5 1,9 10–3
CN– < NO2– < SCN– < N3
– < I– < Br– < Cl– < H2O < NO3–
• Influence des ligands non remplacés trans-[Pt(PEt3)2ACl] + py trans-[Pt(PEt3)2L(py)]+ + Cl–
cis-[Pt(PEt3)2ACl] + py cis-[Pt(PEt3)2L(py)]+ + Cl– A H– CH3
– C6H5– Cl–
constantes de vitesses relatives (isomère trans)
> 104 1700 400 1,0
constantes de vitesses relatives (isomère cis)
3,6 2,3 1,0
Les ligands situés en trans du groupe partant labilisent ce dernier dans l'ordre : F–, H2O, OH–, NH3, py < Cl–< Br– < I– , SCN–, NO2
– < CH3–< H–, PR3, < CO, NO, C2H4, CN–
Deux effets : • effet cinétique des ligands accepteurs π (effet trans) • effet thermodynamique des ligands donneurs σ (influence trans) IV-3 Réactions de substitution dans les complexes octaédriques IV-4-a Echange du solvant [M(H2O)6]z+ + H2O* [M(H2O)5(H2O*)]z+ + H2O La constante de vitesse dépend du métal :
• la réactivité diminue lorsque la charge augmente : [Na(H2O)n]+ > [Mg(H2O)6]2+ > [Al(H2O)6]3+ • elle augmente lorsque le rayon ionique de l'atome central augmente : Mg2+ < Ca2+ < Sr2+ < Ba2+
Zn2+ < Cd2+ < Hg2+ • rôle de l'EACC En champ faible :
n ESCC (Oh) (en unités ∆O) ESCC (C4v) (en unités ΔO) EACC (en unités ΔO) 0, 5, 10 0 0 0
IV-4-b Hydrolyse des complexes de cobalt(III) : type D [CoX(NH3)5]2+ + H2O [Co(NH3)5(H2O)]3+ + X– • influence du groupe partant log k = log K +cte • influence de la charge du complexe : [CoCl2(NH3)4]+ vs. [CoCl(NH3)5]2+. • effets stériques : trans-[CoCl2(N-N)2]+ + H2O trans-[CoCl(H2O)(N-N)2]2+ + Cl–
• Stéréochimie des réactions Proportions des produits d'hydrolyse des complexes cis- et trans-[CoA4BX]n+ et cis- et trans-[Co(AA)2BX]n+ (A : ligand monodentate ; (AA) : ligand bidentate) sur une base statistique :
complexe Intermédiaire
pyramide à base carrée
bipyramide trigonale
% cis % trans % cis % trans trans-[CoA4BX]n+ 0 100 66,6 33,3 cis-[CoA4BX]n+ 100 0 83,3 16,7
• Les ligands accepteurs π diminuent la densité électronique sur M, le rendant ainsi plus sensible à l'attaque par les réactifs nucléophiles. • Les ligands donneurs π en cis de X stabilisent l'intermédiaire pyramide à base carrée; l'interaction π est par contre impossible lorsque le ligand est en trans de X, à moins qu'il n'y ait réarrangement de l'intermédiaire. IV-4-c Remplacement du solvant (anation) Ces réactions sont le plus souvent du type interchange. Exemple 1 : [Co(NH3)5(H2O)]3+ + Yn– → [CoY(NH3)5](3-n)+ + H2O K[Y] >>1 v = k[{Co(NH3)5(H2O)}3+]
Yn– N3– SO4
2– Cl– SCN–
k (s-1, à 45 °C) 10–4 2,4 10–5 2,1 10–5 1,6 10–5 Arguments en faveur d'un mécanisme Id : • mise en évidence de deux étapes lorsque Y2– = SO4
2– : la variation instantanée du spectre UV lors du mélange des réactifs est attribuée à une bande de transfert de charge caractéristique d'une paire d'ions : [Co(NH3)5(H2O)]3+ + SO4
2– {[Co(NH3)5(H2O)]3+,SO42–} K = 2 103 mol–1 l
Elle est suivie d'une évolution beaucoup plus lente de la partie visible du spectre : {[Co(NH3)5(H2O)]3+,SO4
2–} → [Co(SO4)(NH3)5]+ + H2O • k est pratiquement indépendant du groupe entrant. Exemple 2 : [Ni(H2O)6]2+ + Y [NiY(H2O)5]2+ + H2O K[Y] <<1 v = kobs[{Ni(H2O)6}2+][Y] = kK[{Ni(H2O)6}2+][Y]
Y CH3CO2– F– NH3 SCN–
K (mol–1 l+1) 3 1 0,15 1 kobs (mol–1 l s_1) 105 8 103 5 103 6 103
IV-4-d Réactions d'isomérisation • Avec rupture de liaison trans-[Co(en)2Cl2]+ cis-[Co(en)2Cl2]+ mécanisme dissociatif (intermédiaire bipyramide trigonale). • Sans rupture de liaison Δ-[Ni(en)3]2+ Λ-[Ni(en)3]2+ passage par une forme prismatique trigonale.
31
Chapitre V Réactions de transfert d’électron On considère ici les réactions de transfert d'électron entre deux complexes métalliques, le donneur D et l'accepteur A. Deux mécanismes ont été identifiés par Henry Taube : le mécanisme par sphère externe et le mécanisme par sphère interne. Dans le premier, l'état de transition est du type {D,A}‡ : les sphères les sphères de coordination ne s'interpénètrent pas ; dans le second, l'état de transition est de type {D-X-A}‡ où X est un ligand pont entre les deux centres métalliques. • Lorsque la réaction redox est rapide alors que le donneur et l’accepteur sont inertes, on peut affirmer que le mécanisme est du type sphère externe. • Lorsque la réaction redox s'accompagne du transfert d'un ligand entre deux centres métalliques l'un et l'autre inertes, on peut affirmer que le mécanisme est du type sphère interne. • Dans tous les autres cas, la conclusion est en général moins évidente. V-1 Réactions de type sphère externe V-1-a Mécanisme général • Formation du complexe précurseur : D + A {D,A} KSE • Activation du complexe précurseur : {D,A} {D,A}‡ ΔG‡
• Transfert d'électron : {D,A}‡ {D+,A-}‡ kTE
• Relaxation du complexe successeur : {D+,A-}‡ {D+,A-} • Dissociation du complexe successeur : {D+,A-} D+ + A- V-1-b Courbes d'énergie potentielle Référence : R.A. Marcus, Electron Transfer Reactions in Chemistry: Theory and Experiment (Nobel Lecture), Angew. Chem. Int. Ed. Engl. 1993, 32, 1111-1121. L'énergie potentielle des réactifs (A, D et solvant) est fonction d'un très grand nombre de coordonnées, N, incluant les coordonnées nucléaires de A et D, la position et l'orientation des molécules de solvant. Il en est de même de l'énergie potentielle des produits (A-, D+ et solvant). L'intersection des surfaces d'énergie potentielle UP et US est une surface à N-1 dimensions. Il est possible de remplacer cette description par une représentation où l'enthalpie libre du système, G, est fonction quadratique d'une coordonnée de réaction globale, x : G = λx2. Le système {A,D,solvant} est alors représenté par un point situé sur la parabole P. De même, le système {A-
,D+,solvant} est représenté par un point situé sur la parabole S. Lorsque les coordonnées nucléaires des complexes dépendent peu de l'état d'oxydation du métal, les deux paraboles sont très rapprochées ; par contre, elles sont très séparées lorsque les positions des noyaux sont très sensibles à l'état d’oxydation du métal. Le point représentatif du système se déplace sur la parabole P ; lorsqu'il atteint le point d'intersection des deux courbes, il peut y avoir passage de l'une à l'autre, donc transfert d'électron de D vers A. Cette description correspond à un transfert thermiquement activé avec une enthalpie libre d'activation ΔG‡. Cependant, pour que la probabilité de transfert ne soit pas nulle au point d'intersection des deux paraboles, il faut qu'il y ait mélange des états {A,D,solvant} et (A–,D+,solvant}. L'interaction a pour effet de séparer les courbes P et S au voisinage du point d'intersection T.
A.G. Lappin, Redox Mechanisms in Inorganic Chemistry, Ellis Horwood, 1994, p. 86.
La constante de transfert d'électron, kTE, a pour expression : KTE = κνnΓKSEexp(-ΔG‡/RT) =Zexp(-ΔG‡/RT) où KSE désigne la constante d'équilibre de formation du complexe précurseur, κ dépend de l'interaction entre le donneur et l'accepteur, νn désigne la fréquence nucléaire effective, principalement déterminée par les fréquences de vibration des sphères de coordination du donneur et de l'accepteur (νn est de l'ordre de 1013 s-1), Γ représente un terme correctif d'effet tunnel, d'origine quantique.
L'enthalpie d'activation, ΔG‡, se compose de deux termes : l'énergie de réorganisation des sphères de coordination (ΔG‡
int) et l'énergie de réorganisation des sphères de solvatation (ΔG‡iext).
• !Gint
‡ = 3kA(d - dA)2 + 3kD(d - dD)2
où kA et kD sont les constantes de force et dA et dD sont les longueurs des liaisons métal-ligand, respectivement dans l'accepteur et dans le donneur, dA et dD ; d est la longueur commune des liaisons métal-ligand dans l'état de transition. Pour trouver la valeur de d correspondant au trajet
optimum, on annule !!Gint‡
!d.
!!G
int
‡
!d = 6kA(d - dA) + 6kD(d - dD) = 0
D'où :
d = (kAdA + kDdD)
kA + kD
!Gint
‡ =NA
3kAkD(dA - dD)2
kA + kD
• !Gext
‡ = NA
(!ze)2
16"#0
[ 12aA
+ 12aD
- 1r][ 1
n2 - 1#r
]
où Δz est le nombre d'électrons transférés au cours de la réaction ; aA et aD sont les rayons des sphères de van der Waals de l'accepteur et du donneur ; r est la distance entre les centres des réactifs, n est l'indice de réfraction du milieu, εr sa constante diélectrique (permittivité relative). Exemples : Ox/Red kTE, M-1 s-1 Δd (Å) r (Å) [Fe(H2O)6]3+/[Fe(H2O)6]2+ t2g
Pour le couple CoIII/CoII, le transfert d’électron nécessite l’activation préalable du cobalt(III) et du cobalt(II). (D’autres chemins
peuvent être envisagés).
V-1-d Réactions hétéronucléaires – Relation de Marcus On considère les réactions homonucléaires symétriques : [M1(H2O)6]2+ + [*M1(H2O)6]3+ [*M1(H2O)6]2+ + [M1(H2O)6]3+ (11)
[M2(H2O)6]2+ + [*M2(H2O)6]3+ [*M2(H2O)6]2+ + [M2(H2O)6]3+ (22) et la réaction croisée : [M1(H2O)6]2+ + [M2(H2O)6]3+ [M1(H2O)6]3+ + [M2(H2O)6]2+ (12)
A.G. Lappin, Redox Mechanisms in Inorganic Chemistry, Ellis Horwood, 1994, p. 53.
Les courbes décrivant l'état initial et l'état final du système lors de la réaction croisée ont respectivement pour équations : ΔGP = λ12 x2 et ΔGS = λ12(1-x)2 + ΔG0
34 L'intersection des deux courbes a pour coordonnées :
x‡ =
!12 + "G0
2!12
!G‡ = "12x‡2 =
("12 + !G0)2
4"12 La relation précédente peut être réécrite comme suit :
!G‡ =
"12
4 + !G0
2 +
(!G0)2
4"12 Si l'on admet que le facteur pré-exponentiel Z est le même pour les trois réactions et que l'énergie de réorganisation de la réaction croisée, λ12, est égale à la moyenne arithmétique des énergies de réorganisation des réactions symétriques, respectivement λ11 et λ22, on obtient :
!G12
‡ = !G11
‡ + !G22
‡ + !G
0
2 +
(!G0)2
4"12
!G12
‡ = 1
2(!G11
‡ + !G22
‡ + !G0) + 1
2[
!G0 2
4(!G11‡
+ !G22‡
)
]
On pose :
[
!G0 2
4(!G11‡
+ !G22‡
)
] = -RT Ln f12
Compte tenu des relations :
kij = Zexp[- !Gij
‡
RT]
K12 = exp[- !G0
RT]
on en déduit la relation de Marcus : k12 = [k11k22K12f12]1/2 avec :
ln f12 = (ln K12)2
4 ln k11k22
Z2 Remarque : dans beaucoup d'applications, on peut prendre f12 = 1. On distingue deux régions : • la région normale (-∆G0/λ < 1) : lorsque ∆G0 augmente en valeur absolue, l’énergie d’activation décroît et s’annule lorsque ∆G0 = -λ. • la région inverse (-∆G0/λ > 1) : l’énergie d’activation croît lorsque ∆G0 augmente (en valeur absolue).
35
A.G. Lappin, Redox Mechanisms in Inorganic Chemistry, Ellis Horwood, 1994, p. 54.
V-2 Mécanisme de type sphère interne V-2-a Mécanisme général • Formation du complexe précurseur : [L5M1
III—X] + [Y—M2IIL’5] [L5M1
III—X—M2IIL’5] + Y
• Transfert d'électron : [L5M1
III—X—M2IIL’5] [L5M1
II—X—M2IIIL’5]
• Dissociation du complexe successeur : [L5M1
II—X—M2IIIL’5] {L5M1
II} + [X–M2IIIL’5]
{L5M1II} ...
ou : [L5M1
II—X—M2IIIL’5] [L5M1
II—X] + {M2IIIL’
5} {M2
IIIL’5} ...
Remarques : i) Chacune de ces étapes requiert une activation ; chacune d'elle peut constituer l'étape limitante de la réaction globale ; ii) Le complexe [Y–M2
IIL’5] doit être labile et le ligand X doit être apte à jouer le rôle de pont; iii) Dans le complexe successeur, la coupure s'effectue du côté du centre le plus labile. Type I : le transfert électronique constitue l'étape déterminante. Exemple : [CoCl(NH3)5]2+ + [Cr(H2O)6]2+ + 5H3O+ [Co(H2O)6]2+ + [CrCl(H2O)5]2+ + 5NH4
+
k = 6 105 mol-1 l s-1 à 25 °C et µ = 0,1 M. [(NH3)5CoIII-Cl]2+ + [CrII(H2O)6]2+ [(NH3)5CoIII-Cl-CrII(H2O)5]4+ rapide [(NH3)5CoIII-Cl-CrII(H2O)5]4+ [(NH3)5CoII-Cl-CrIII(H2O)5]4+ limitante
+ rapide Type II : la formation du complexe précurseur constitue l'étape déterminante. Exemple : [CoX(NH3)5]2+ + [V(H2O)6]2+ + 5H3O+ + H2O [Co(H2O)6]2+ + [V(H2O)6]3+ + 5NH4
+ + X-
X- = NCS- k = 28 mol-1 l s-1 X- = N3
- k = 13 mol-1 l s-1 ΔH‡ = 49,0 kJ mol-1 ΔS‡ = - 58 J K-1 mol-1
On peut comparer ces paramètres à ceux de la réaction : [V(H2O)6]2+ + X- [VX(H2O)5]+ + H2O X- = NCS- k = 28 mol-1 l s-1 ΔH‡ = 56,5 kJ mol-1 ΔS‡ = - 29 J K-1 mol-1
La réaction de substitution sur le vanadium, relativement inerte, constitue l'étape limitante de la réaction. Type III : la dissociation du complexe successeur constitue l'étape déterminante Exemple : [RuIIICl(NH3)5]2+ + [CrII(H2O)6]2+ [(NH3)5RuII—Cl—CrIII(H2O)5]4+ équilibrée [(NH3)5RuII—Cl—CrIII(H2O)5]4+
V-2-b Critères diagnostiques du mécanisme La labilité de [Cr(H2O)6]2+ et la lenteur de l’auto-échange sphère externe du couple [Cr(H2O)6]3+/[Cr(H2O)6]2+ font que les réactions de réduction par [Cr(H2O)6]2+ sont en général de type sphère interne, du moins lorsque l’accepteur possède un ligand susceptible de jouer le rôle de pont. Pour les autres réducteurs ([V(H2O)6]2+, [Fe(H2O)6]2+, [Ti(H2O)6]3+…) il n’est pas toujours facile de déterminer le mécanisme. Certaines comparaisons portant sur les ligands susceptibles de jouer le rôle de pont peuvent alors se révéler utiles : une faible réactivité de H2O par comparaison avec OH-, ou une forte réactivité de N3
- par comparaison avec SCN- sont en faveur d’un mécanisme sphère interne. On peut aussi noter que le mécanisme sphère interne est favorisé lorsqu’au moins l’une des orbitales concernées par le transfert d’électron est de type σ.
Réactifs Orbitales de l'accepteur et du donneur
KSI/kSE
V(III)/V(II) π/π 10 V(III)/Cr(II) π/σ 104
Cr(III)/Cr(II) σ/σ 107
Fe(III)/Fe(II) π/π 10 V-2-c Rôle du ligand pont Deux mécanismes peuvent être distingués : le mécanisme résonant et le mécanisme chimique. • Dans le mécanisme résonant, l'état de transition est décrit à l'aide d'un modèle orbitalaire tricentrique impliquant trois orbitales de type σ, l'une sur le donneur, l'autre sur le pont, l'autre sur l'accepteur. Ce modèle s'applique aux ligands non réductibles et en particulier aux ligands à couche pleine, e.g. X-, O2-, N3-... • Dans le mécanisme chimique, l'électron transite par le pont. Il ne peut intervenir que si le pont dispose d'orbitales vacantes accessibles, i.e. s'il est réductible. C'est, par exemple, le cas du ligand isonicotinamide (E = - 0,41 V/ECS dans HClO4 1M à 25 °C). Ce mécanisme peut être décrit de la façon suivante : [L5MIII—X]3+ + [Cr(H2O)6]2+ [L5MIII—Xred–CrIII(H2O)5]5+ + H2O (k1, k-1)
37
[L5MIII—Xred–CrIII(H2O)5]5+ produits (k2) La vitesse de formation des produits est alors donnée par l'expression : v = k1k2
k-1 + k2
[L5MIII—X][Cr2+]
Exemple : réduction de divers oxydants par [Cr(H2O)6]2+ Oxydant K (mol-1 l+1 s-1 M (mol.L-1) Mécanisme [(NH3)5Co-Cl]2+ 6,0 105 0,1 ”résonant” [(NH3)5Co-isonocotinamide]3+ 17,4 1 ”chimique” [(H2O)5Cr-Cl]2+ 33 1,5 ”résonant” [(H2O)5Cr-isonocotinamide]3+ 1,8 1 ”chimique” [(NH3)5Co-isonocotinamide]3+ 3,92 105 1 ”résonant”
Lorsque k2 est très supérieur à k-1 la vitesse de la réaction est pratiquement insensible à la
nature de l'accepteur. Ainsi, les constantes de vitesse des réactions de réduction des complexes [(NH3)5CoIII-isonicotinamide]3+ et [(H2O)5CrIII-isonicotinamide]3+ par [Cr(H2O)6]2+ sont à peu près les mêmes alors que la réduction des complexes [(NH3)5CoIII-X]2+ (X = F, Cl, OH) par [Cr(H2O)6]2+ est environ 105 fois plus rapide que celle des complexes [(H2O)5CrIII-X]2+. L'étape limitante est alors la réduction du ligand isonicotinamide. En revanche la réduction du complexe [(NH3)5RuIII-X]3+ est beaucoup plus rapide. La différence s’explique si l'on considère la symétrie des orbitales des accepteurs : σ* pour CoIII et CrIII (mauvais recouvrement avec les orbitales π centrées sur le ligand isonicotinamide), π pour RuIII (bon recouvrement, l’électron ne reste pas sur le ligand). V-2-d Applications : voir chapitre VI
38
Chapitre VI – Synthèse des composés de coordination
VI-1 Réactions de substitution et/ou de transfert d’électron(s) VI-1-a Synthèse des complexes de platine(II) Les séquences ci-dessous s’expliquent par l’effet trans (NH3 < py< Cl- < Br-, cf chapitre IV) et par la stabilité relative des liaisons (Pt—NH3 > Pt—Br > Pt—Cl).
VI-1-b Exemples divers VI-1-b-α Hydrolyse basique des complexes de cobalt(III)
Les réactions de substitution des complexes de Co(III) sont accélérées en milieu alcalin : [CoCl(NH3)5]2+ + OH– →[Co(OH)(NH3)5]2+ + Cl– v = k1K[{CoCl(NH3)5}2+][OH–] Mécanisme : [CoCl(NH3)5]2+ + OH– [Co(NH3)4(NH2)Cl]+ + H2O (rapide, équilibrée, constante K) [Co(NH3)4(NH2)Cl]+ → [Co(NH3)4(NH2)]2+ + Cl– (lente, k1) [Co(NH3)4(NH2)]2+ + H2O → [Co(OH)(NH3)5]2+ (rapide)
VI-1-b-β Orientation des réactions de substitution [CoCl(NH3)5]2+ + CN- → [Co(CN)(NH3)5]2+ + Cl- en l'absence de Co2+
[CoCl(NH3)5]2+ + 5CN- → [CoCl(CN)5]3- + 5NH3 en présence de Co2+
L'attaque par Cr2+ se fait sensiblement à la même vitesse sur l'atome d'azote (attaque ”éloignée”, k = 1,9 105 mol-1 l s-1) et sur l'atome de soufre (attaque ”adjacente”, k = 8 104 mol-1 l s-1) du ligand thiocyanate. Dans le premier cas, on obtient [Cr(NCS)(H2O)5]2+ (forme stable, pourpre), tandis que dans le second on obtient [Cr(SCN)(H2O)5]2+ (forme instable, verte). VI-1-b-δ Catalyse par transfert d'électron
39
Les réactions de substitution sur les complexes de Cr(III), Co(III), Rh(III) et Pt(IV) sont accélérées en présence des états d'oxydation inférieurs, respectivement Cr(II), Co(II), Rh(I) et Pt(II). Exemple : synthèse du complexe [RhCl2(py)4]+
En solution aqueuse, Na3[RhCl6] réagit avec la pyridine mais la réaction s'arrête à la formation de [RhCl3(py)3], insoluble. Il faut chauffer très longtemps à reflux pour obtenir le complexe [RhCl2(py)4]+ alors que ce dernier est obtenu quantitativement et instantanément à température ordinaire si l'on ajoute une petite quantité d'éthanol. Le mécanisme est le suivant :
K.F. Purcell, J. C. Kotz, Inorganic Chemistry, Saunders, 1977, pp. 686-687. VI-2 Réactions des ligands coordonnés – Effet template – Auto-assemblage VI-2-a Activation vs. stabilization VI-2-a-α Exaltation de l’acidité des ligands • Exemple des ammines Complexe pKa Complexe pKa
[Os(NH3)6]3+ > 15 [Os(NH3)6]4+ < 0 trans-[OsCl2(NH3)4]2+ 4,0 cis-[OsCl2(NH3)4]2+ ~ 1 Application à la synthèse des complexes nitrido [Os(NH3)6]3+ + 3Ce4+ + H2O trans-[OsN(H2O)(NH3)4]3+ + 3Ce3+ + NH4
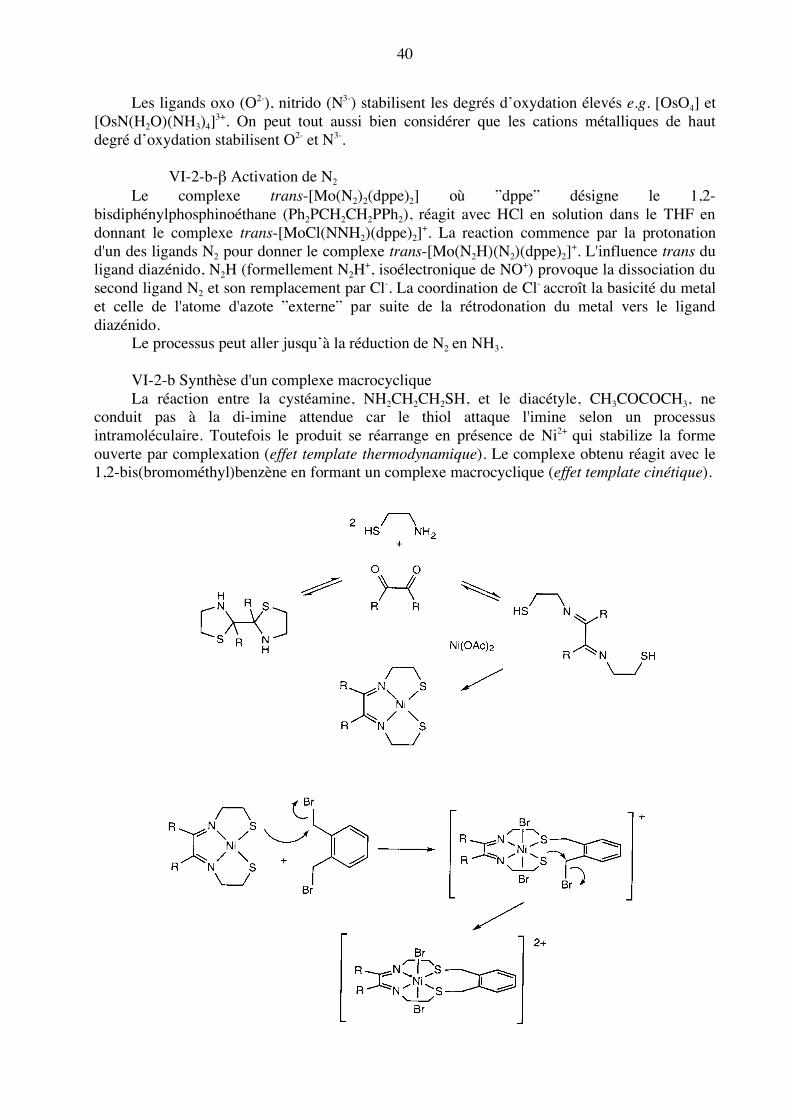
Les ligands oxo (O2-), nitrido (N3-) stabilisent les degrés d’oxydation élevés e.g. [OsO4] et [OsN(H2O)(NH3)4]3+. On peut tout aussi bien considérer que les cations métalliques de haut degré d’oxydation stabilisent O2- et N3-. VI-2-b-β Activation de N2 Le complexe trans-[Mo(N2)2(dppe)2] où ”dppe” désigne le 1,2-bisdiphénylphosphinoéthane (Ph2PCH2CH2PPh2), réagit avec HCl en solution dans le THF en donnant le complexe trans-[MoCl(NNH2)(dppe)2]+. La reaction commence par la protonation d'un des ligands N2 pour donner le complexe trans-[Mo(N2H)(N2)(dppe)2]+. L'influence trans du ligand diazénido, N2H (formellement N2H+, isoélectronique de NO+) provoque la dissociation du second ligand N2 et son remplacement par Cl-. La coordination de Cl- accroît la basicité du metal et celle de l'atome d'azote ”externe” par suite de la rétrodonation du metal vers le ligand diazénido.
Le processus peut aller jusqu’à la réduction de N2 en NH3. VI-2-b Synthèse d'un complexe macrocyclique La réaction entre la cystéamine, NH2CH2CH2SH, et le diacétyle, CH3COCOCH3, ne conduit pas à la di-imine attendue car le thiol attaque l'imine selon un processus intramoléculaire. Toutefois le produit se réarrange en présence de Ni2+ qui stabilize la forme ouverte par complexation (effet template thermodynamique). Le complexe obtenu réagit avec le 1,2-bis(bromométhyl)benzène en formant un complexe macrocyclique (effet template cinétique).
41
VI-2-c Formation de cages macrobicycliques (”sepulchrates”)
En présence d'une base, le complexe [Co(en)3]3+ réagit avec le nitrométhane et le formaldéhyde : 2,5 g de [Co(en)3]Br3 et 1,5 g de Na2CO3 sont dissous dans 90 mL d'eau ; on ajoute 5 mL de nitrométhane et 40 mL d'une solution aqueuse à 37% de formaldéhyde. La solution est placée au bain-marie à 40 °C. La couleur de la solution passe rapidement du jaune au brun et des cristaux jaunes du composé [Co(dinosar)]Br3 apparaissent au bout d'une heure (dinosar = dinitrosarcophagine).
Le mécanisme est le suivant : [Co(en)3]3+ réagit avec le formaldéhyde en donnant un aminol qui se déshydrate en formant une imine. L'imine coordonnée est attaquée par la base conjuguée du nitrométhane pour donner un intermédiaire qui réagit à son tour en formant une imine, laquelle subit une attaque nucléophile intramoléculaire. Le processus se répète …
L'environnement du cobalt est octaédrique. La réaction est stéréospécifique : si l'on part du complexe ∆-[Co(en)3]3+ on obtient le complexe ∆-[Co(dinosar)]3+. L'encapsulation du métal le rend inerte, même après réduction, alors que Co(II) est normalement labile. Ainsi, la réduction du composé [CoIII(dinosar)]Br3 par le zinc en présence d'acide chlorhydrique suivie d'une réoxydation l'air conduit au composé [CoIII(diamsarH2]Cl5 (diamsar = diaminosarcophagine).
J.MacB Harrowfield, G.A. Lawrance, A.M. Sargeson, J. Chem. Educ., 1985, 62, 804-806.
42
VI-2-d Auto-assemblage VI-2-d-α Carrés et cages octaédriques
• Le complexe [Pd(en)(ONO2)2] et la 4,4'-bipyridine réagissent en formant un carré de stoechiométrie M4L4. • Le complexe [Pd(en)(ONO2)2] et le ligand 2,4,6-tris(4-pyridyl)1,3,5-triazine réagissent en formant un assemblage de stoechiométrie M4L6 et de symétrie Td.
M. Fujita et al., Chem. Commun., 2001, 509-518; Acc. Chem. Res., 2005, 38, 371-380. VI-2-d-β Caténanes
En solution aqueuse, le complexe 1a (M = Pt) est stable, mais il donne le complexe 2a après addition de nitrate de sodium ([NO3]- = 5 mol l-1) et chauffage à 100 °C. Le complexe 2a, isolé sous forme de perchlorate et redissous dans l’eau est stable, même à 100°C. M. Fujita, F. Ibukuro, K. Yamaguchi, K. Ogura, J. Am. Chem. Soc., 1995, 117, 4175-4176.