ORGANICK ORGANICK Á Á CHEMIE II CHEMIE II Zkratky: Me = methyl; Et = ethyl; Bu = (n)-butyl; Ph = fenyl; Ar = aryl; Cp = cyklopentadienyl; Cp* = pentamethylcyklopentadienyl; Ac = acetyl; acac = pentan-2,4-dionát; L = obecný ligand; M = atom kovu obecně (metal) UNIVERZITA JANA EVANGELISTY PURKYNĚ V ÚSTÍ NAD LABEM PŘÍRODOVĚDECKÁ FAKULTA – KATEDRA CHEMIE Doc. Ing. Jan Čermák CSc. 2014
Transcript
ORGANICKORGANICKÁÁ CHEMIE II CHEMIE II
Zkratky: Me = methyl; Et = ethyl; Bu = (n)-butyl; Ph = fenyl; Ar = aryl; Cp = cyklopentadienyl; Cp* = pentamethylcyklopentadienyl; Ac = acetyl; acac = pentan-2,4-dionát; L = obecný ligand; M = atom kovu obecně (metal)
UNIVERZITA JANA EVANGELISTY PURKYNĚ V ÚSTÍ NAD LABEM
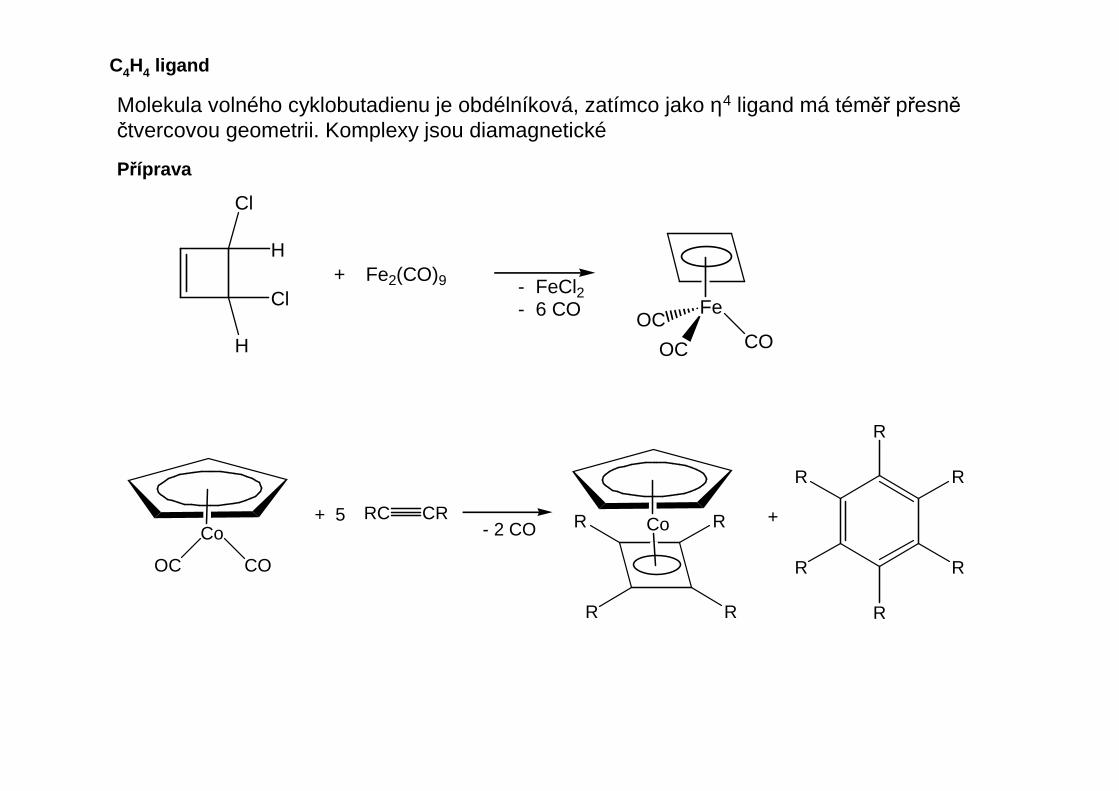
Termodynamické faktory: „stálá“ versus „nestálá“ sloučeninaKinetické faktory: „inertní“ versus „labilní“ sloučenina - především určují reaktivituvůči vnějším vlivům jako je zvýšená teplota, kyslík a voda
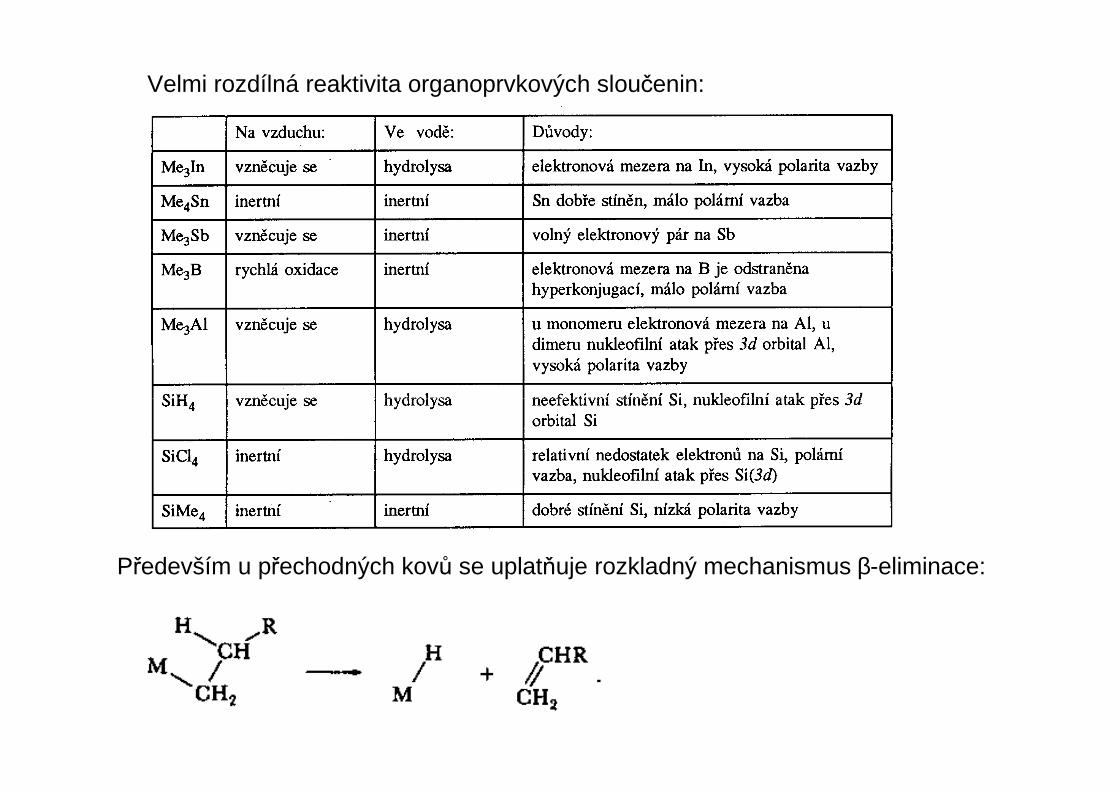
Především u přechodných kovů se uplatňuje rozkladný mechanismus β-eliminace:
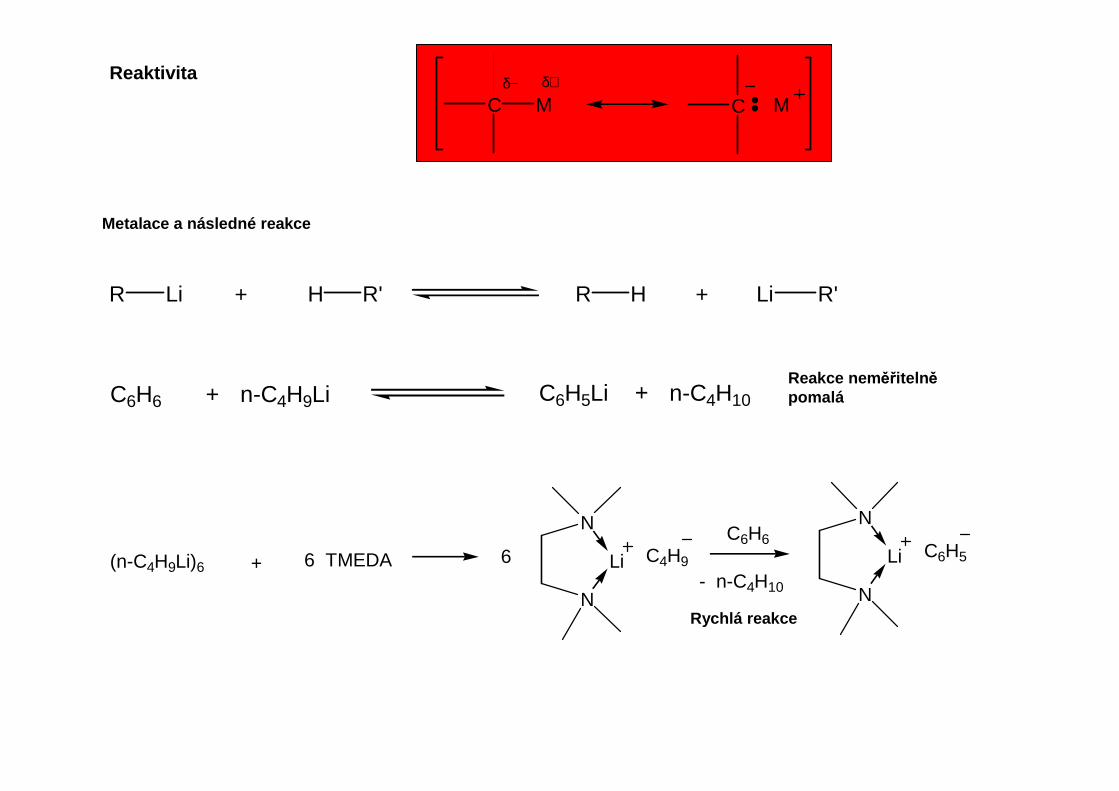
Velmi rozdílná reaktivita organoprvkových sloučenin:
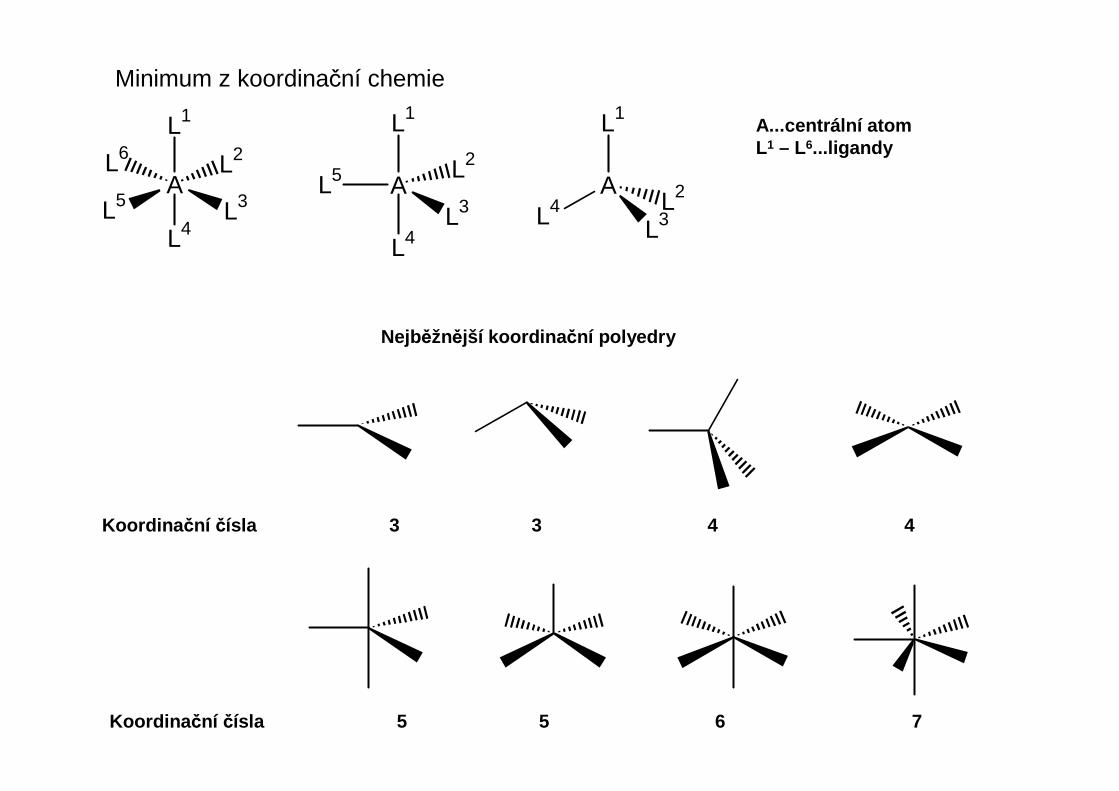
Minimum z koordinační chemie
L3A
L2L6
L5
L1
L4 L4
L1
A
L3L2L5 A
L3
L2
L1
L4
A...centrální atomL1 – L6...ligandy
Nejběžnější koordina ční polyedry
Koordina ční čísla 3 3 4 4
Koordina ční čísla 5 5 6 7
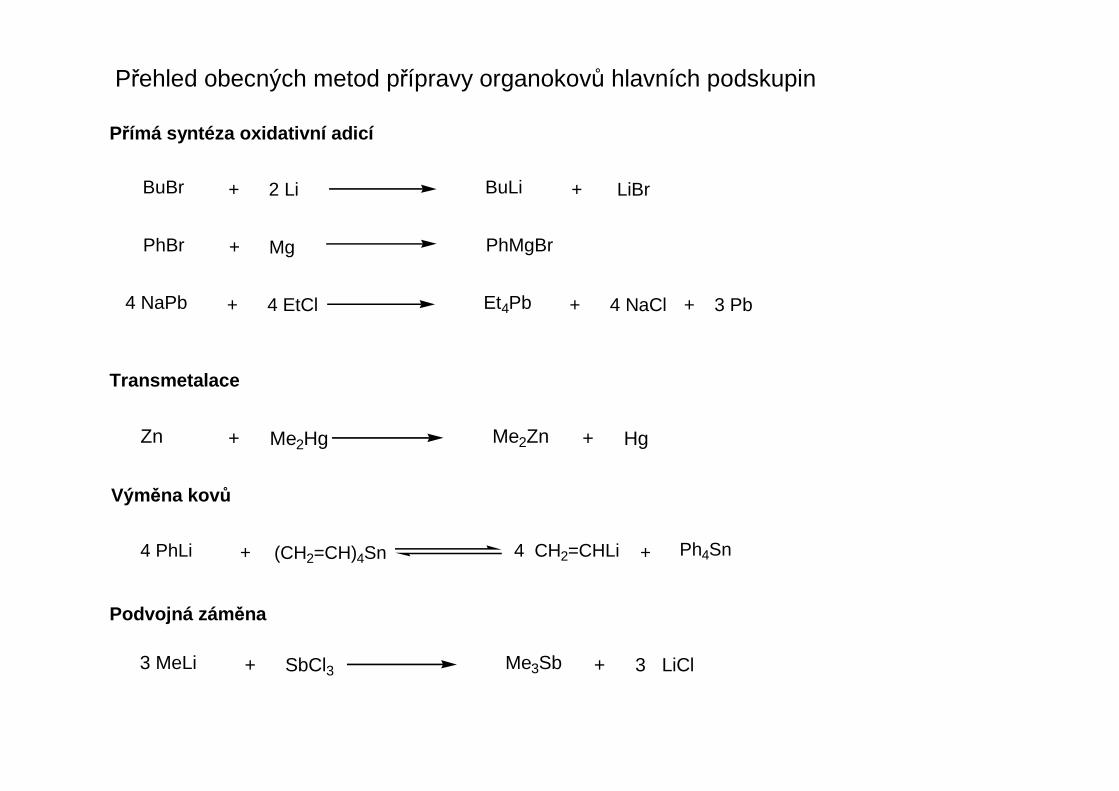
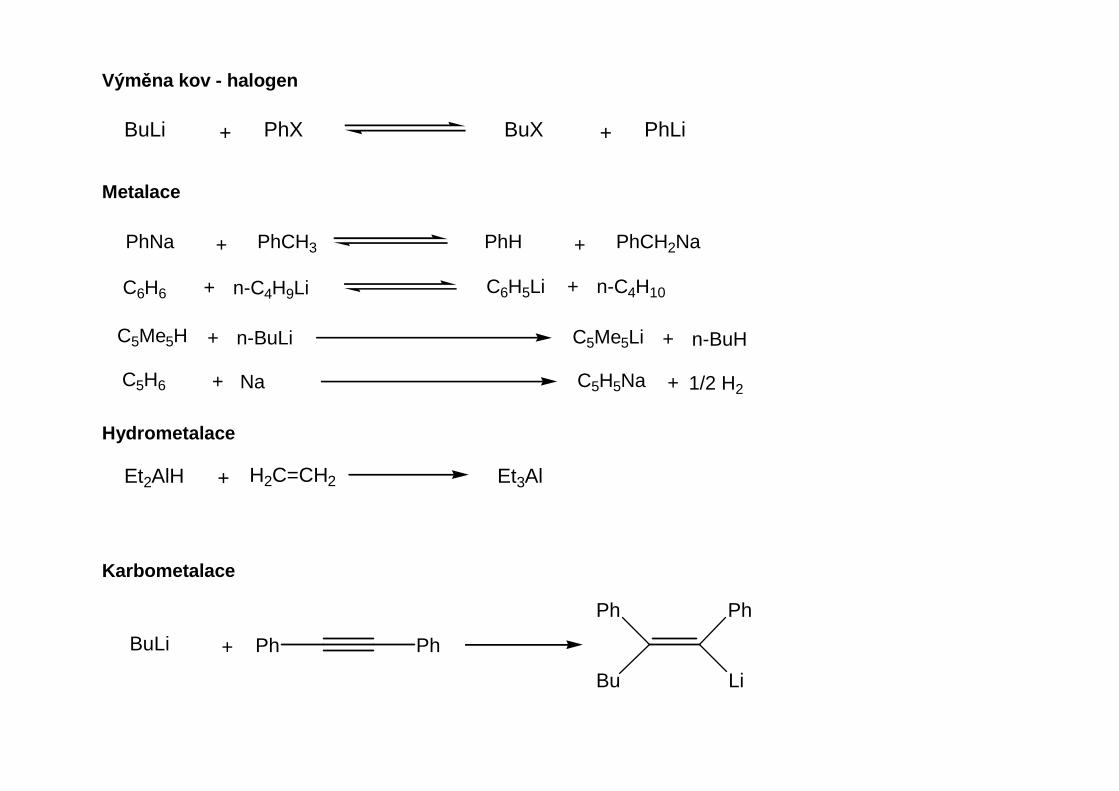
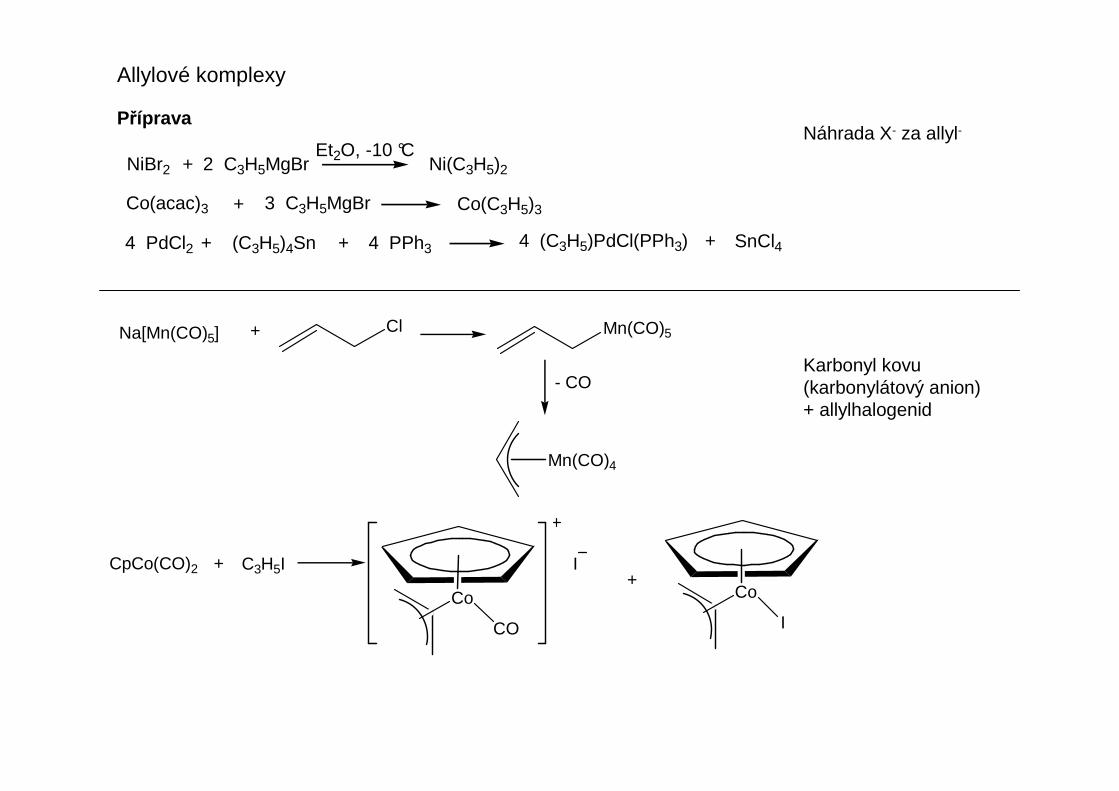
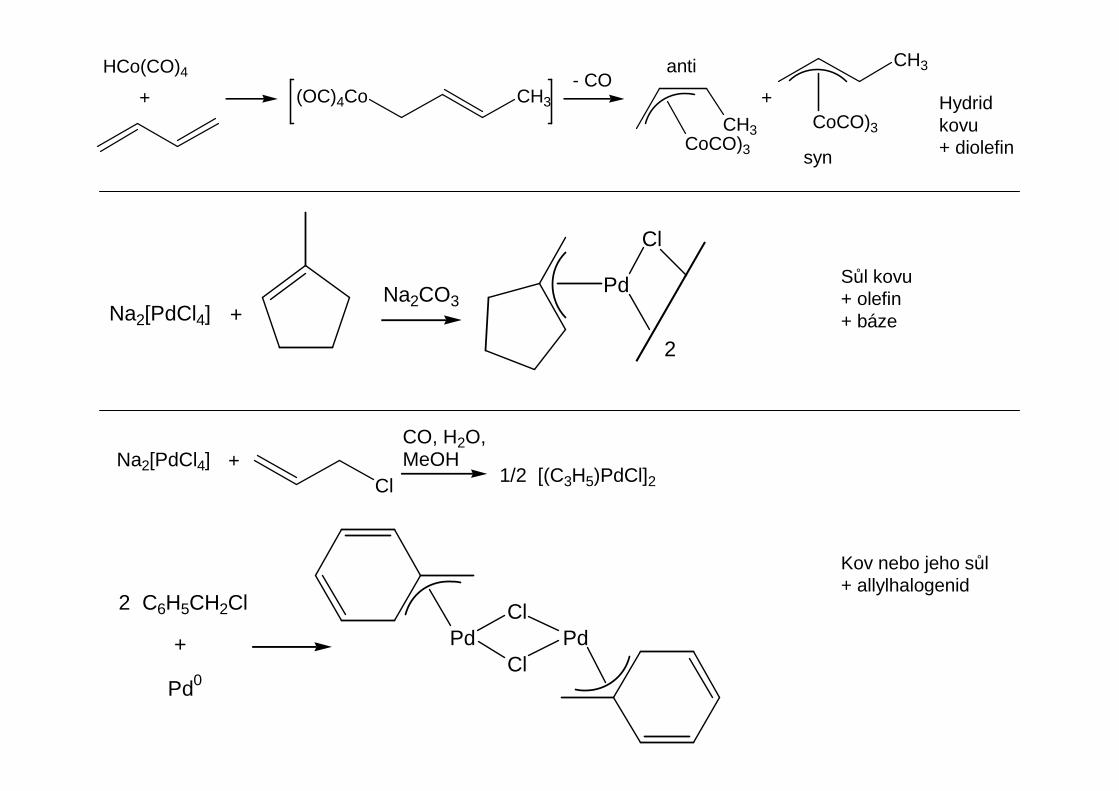
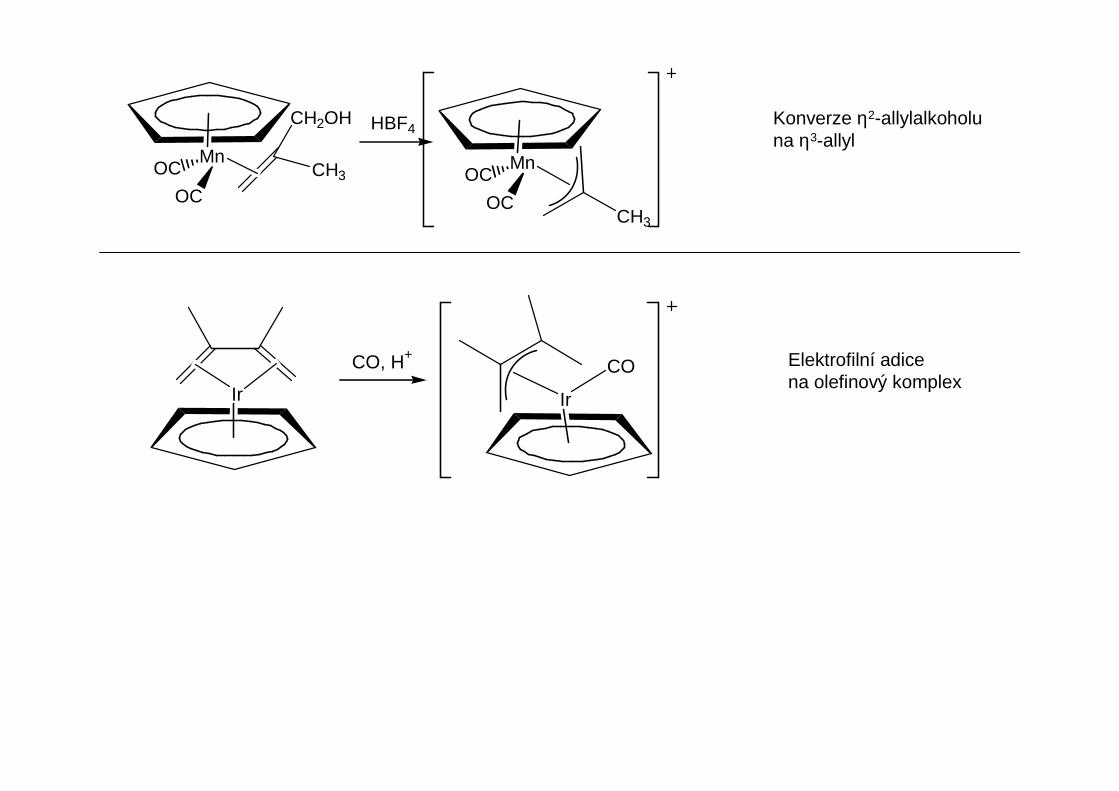
Přehled obecných metod přípravy organokovů hlavních podskupin
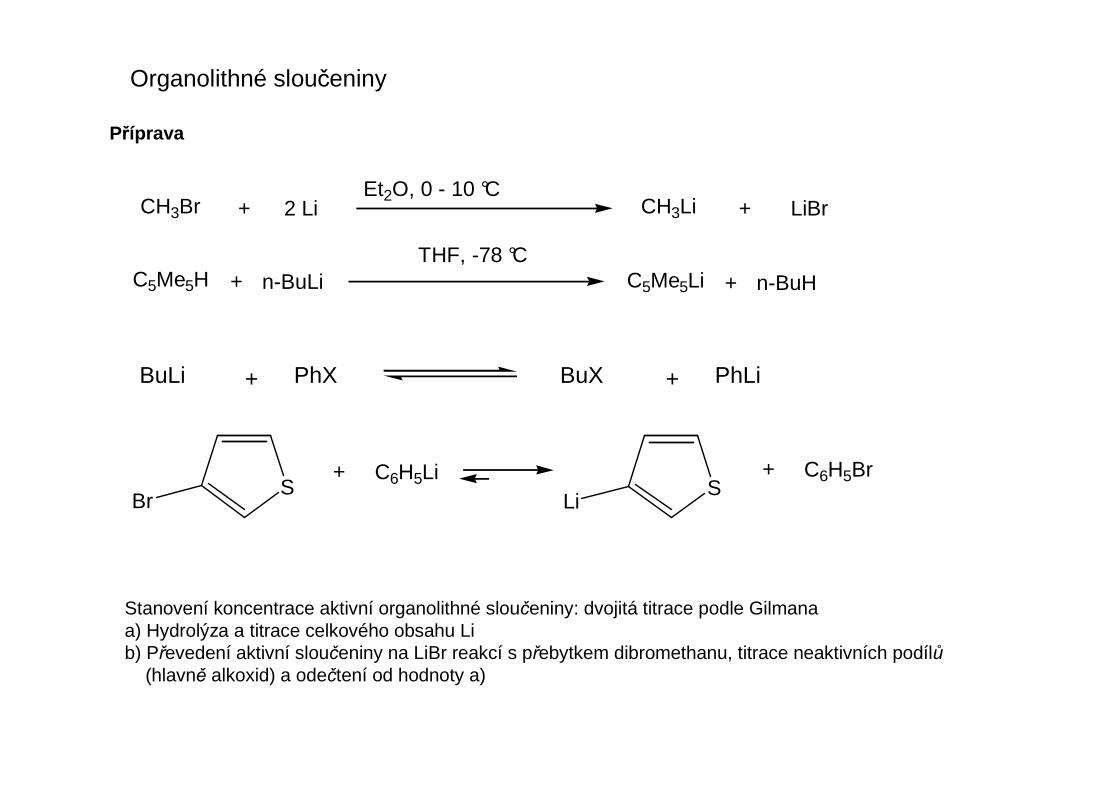
Stanovení koncentrace aktivní organolithné sloučeniny: dvojitá titrace podle Gilmanaa) Hydrolýza a titrace celkového obsahu Lib) Převedení aktivní sloučeniny na LiBr reakcí s přebytkem dibromethanu, titrace neaktivních podílů
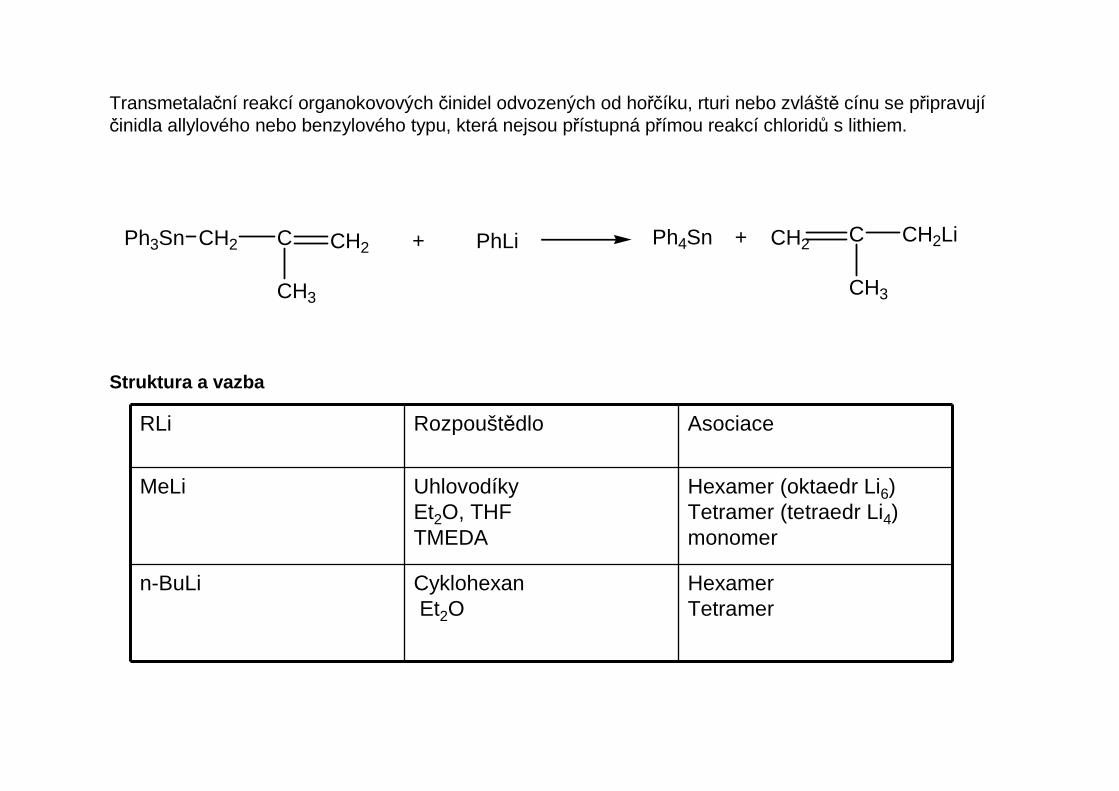
Transmetalační reakcí organokovových činidel odvozených od hořčíku, rturi nebo zvláště cínu se připravujíčinidla allylového nebo benzylového typu, která nejsou přístupná přímou reakcí chloridů s lithiem.
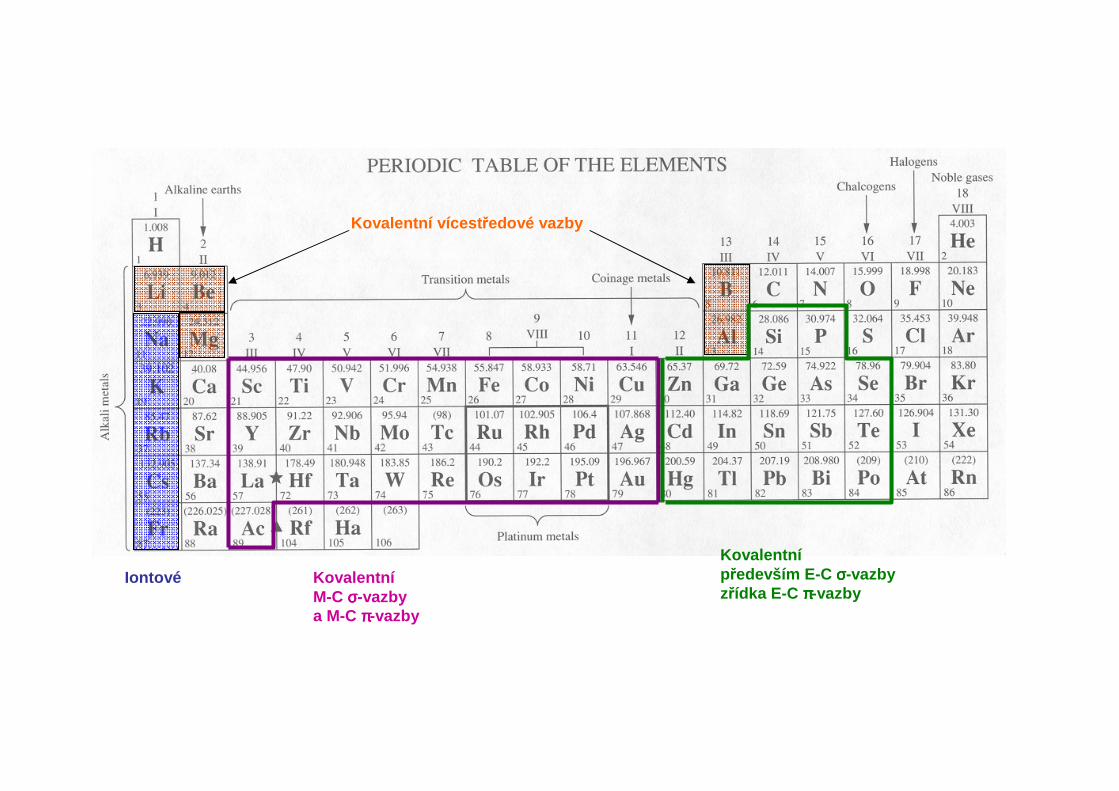
Iontový charakter vazeb M-C vzrůstá od lithia k cesiu. Iontové organické sloučeniny těžších alkalických kovůjsou netěkavé a mimořádně reaktivní, například štěpí ethery:
CH3CH2OEt + KBu- HBu
+K+[C-(CH3)HOEt KOEt H2C=CH2
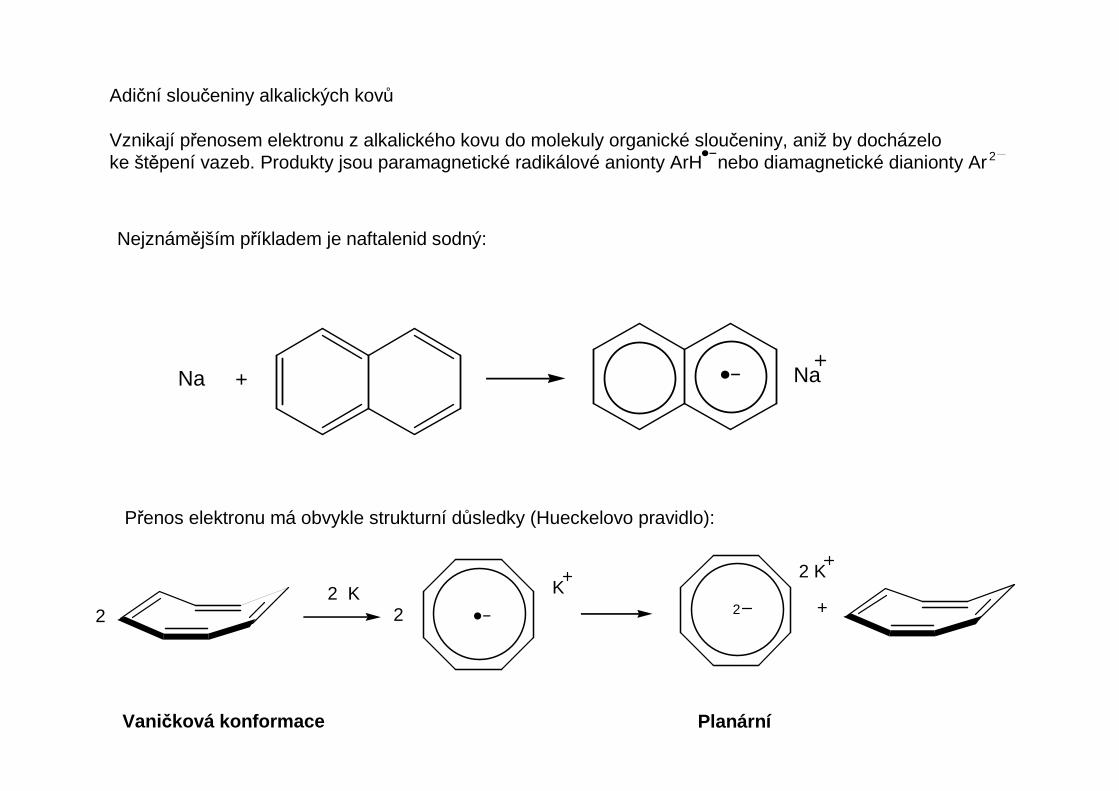
Adiční sloučeniny alkalických kovů
Vznikají přenosem elektronu z alkalického kovu do molekuly organické sloučeniny, aniž by docházeloke štěpení vazeb. Produkty jsou paramagnetické radikálové anionty ArH nebo diamagnetické dianionty Ar 2
Nejznámějším příkladem je naftalenid sodný:
Přenos elektronu má obvykle strukturní důsledky (Hueckelovo pravidlo):
Na + Na
Vaničková konformace Planární
22 K K
2 2 +
2 K
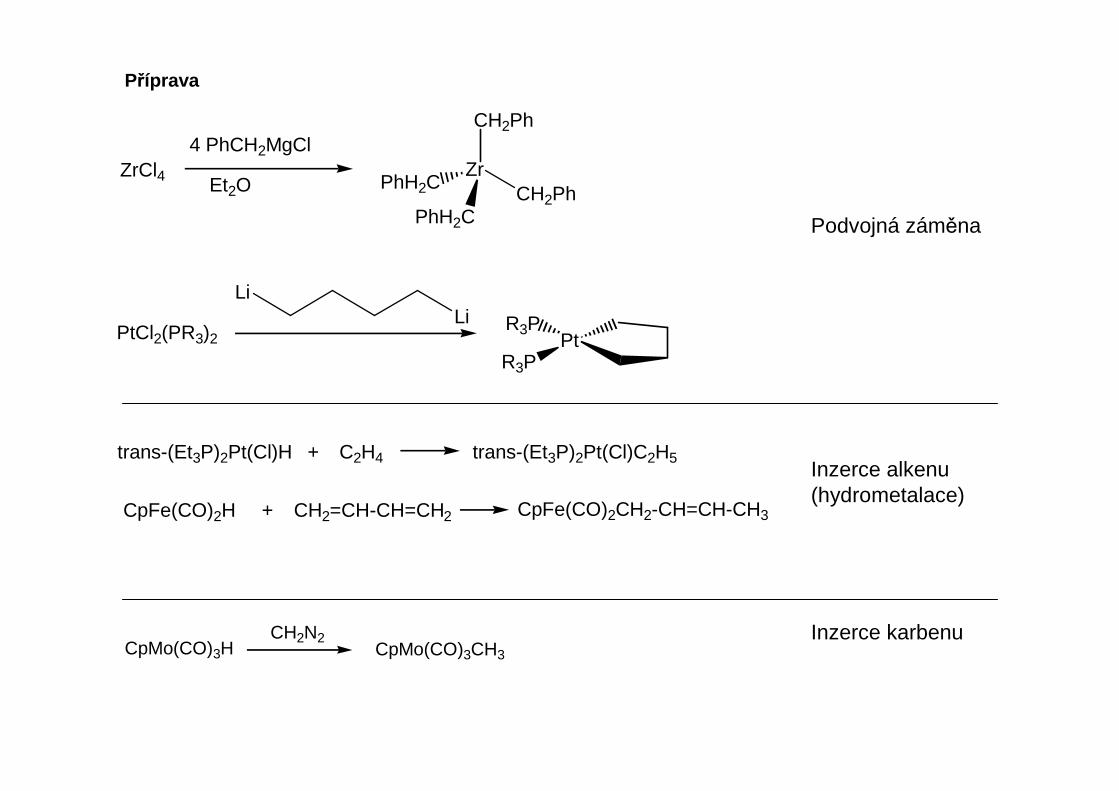
Organohořečnaté sloučeniny
RX + Mg R(CH3CH2)2O
Mg X
CH3CH2OCH2CH3
CH3CH2OCH2CH3
Přítomnost alespoň stechiometrického množství etheru (nejčastěji Et2O nebo THF) je nutná: reakcilze však provést i v jiných rozpouštědlech například alkanech s příměsí THF.Kestabilizaci Grignardových činidel však lze využít i terciární aminy – dimethylanilin, pyridin.
Příprava
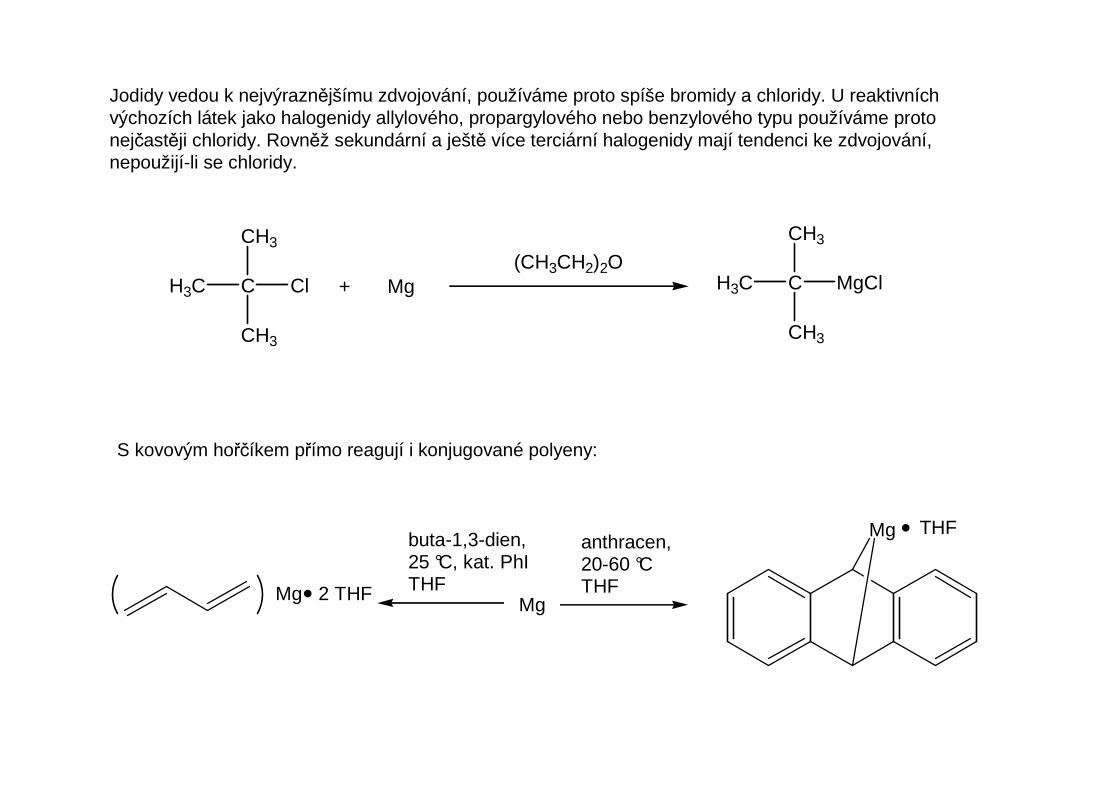
Vliv druhu halogenu a struktury haloalkanu na pr ůběh přímé syntézy
Nejsnáze reagují bromidy a jodidy, nejméně ochotně chloridy, vazba C-F je za podmínek přípravy činidelstabilní. Nežádoucí vedlejší reakcí, v některých případech přímo znemožňující přípravu činidla, jeWurtzova reakce – zdvojení organického zbytku:
2 RX + Mg(CH3CH2)2O
R-R + MgX2
S kovovým hořčíkem přímo reagují i konjugované polyeny:
Mg
anthracen, 20-60 °CTHF
buta-1,3-dien, 25 °C, kat. PhITHF
Mg THF
Mg 2 THF
Jodidy vedou k nejvýraznějšímu zdvojování, používáme proto spíše bromidy a chloridy. U reaktivníchvýchozích látek jako halogenidy allylového, propargylového nebo benzylového typu používáme protonejčastěji chloridy. Rovněž sekundární a ještě více terciární halogenidy mají tendenci ke zdvojování,nepoužijí-li se chloridy.
CH3
CH3C
CH3
Cl + Mg(CH3CH2)2O
CH3
CH3C
CH3
MgCl
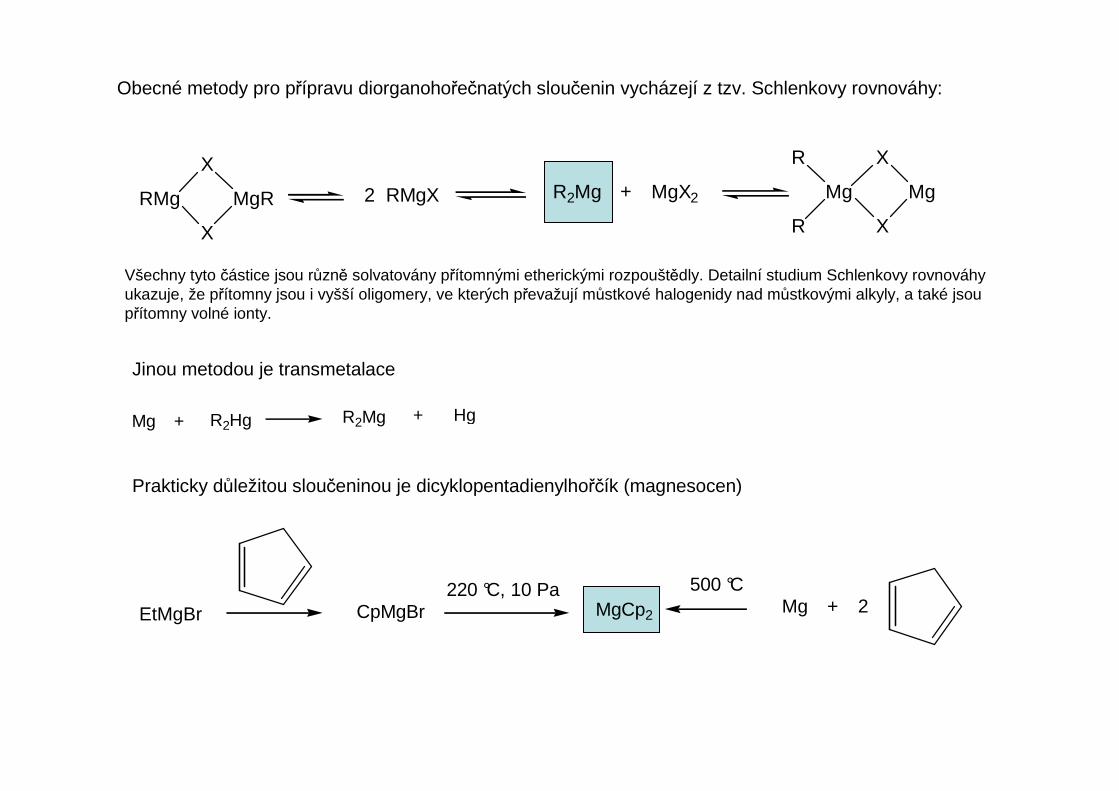
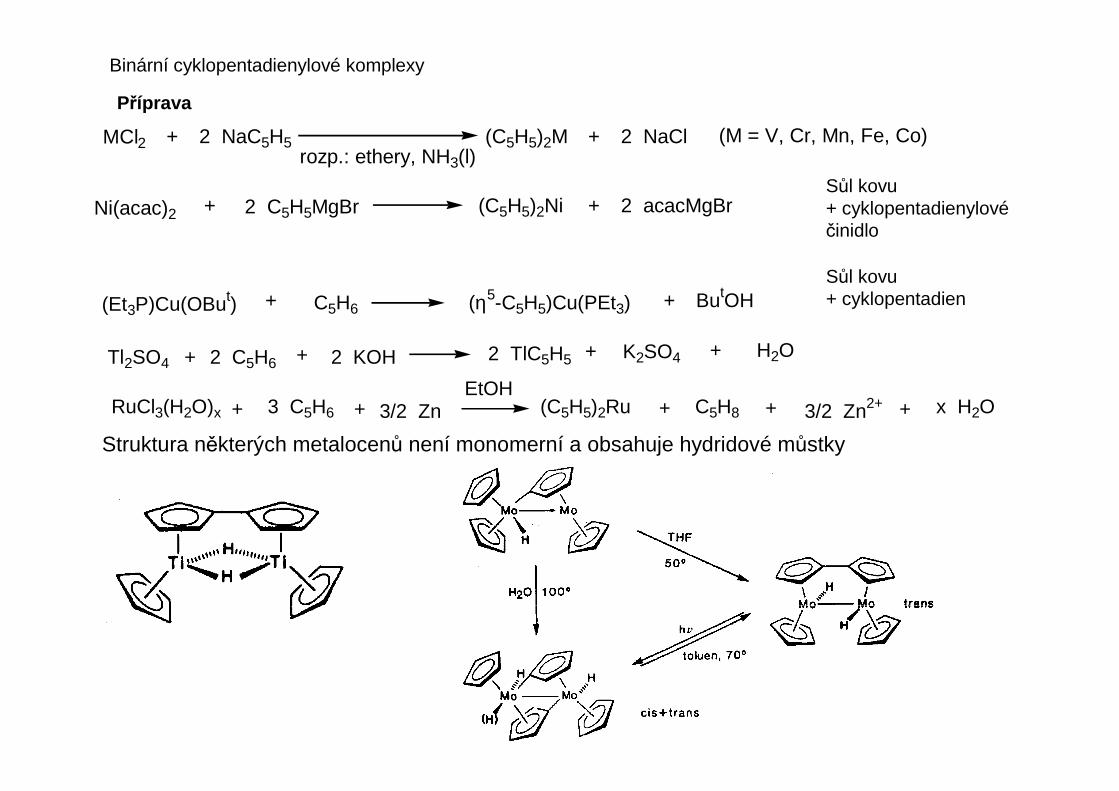
Obecné metody pro přípravu diorganohořečnatých sloučenin vycházejí z tzv. Schlenkovy rovnováhy:
RMg
X
X
MgR 2 RMgX R2Mg + MgX2 Mg
X
X
Mg
R
R
Všechny tyto částice jsou různě solvatovány přítomnými etherickými rozpouštědly. Detailní studium Schlenkovy rovnováhyukazuje, že přítomny jsou i vyšší oligomery, ve kterých převažují můstkové halogenidy nad můstkovými alkyly, a také jsoupřítomny volné ionty.
Jinou metodou je transmetalace
Mg + R2Hg R2Mg + Hg
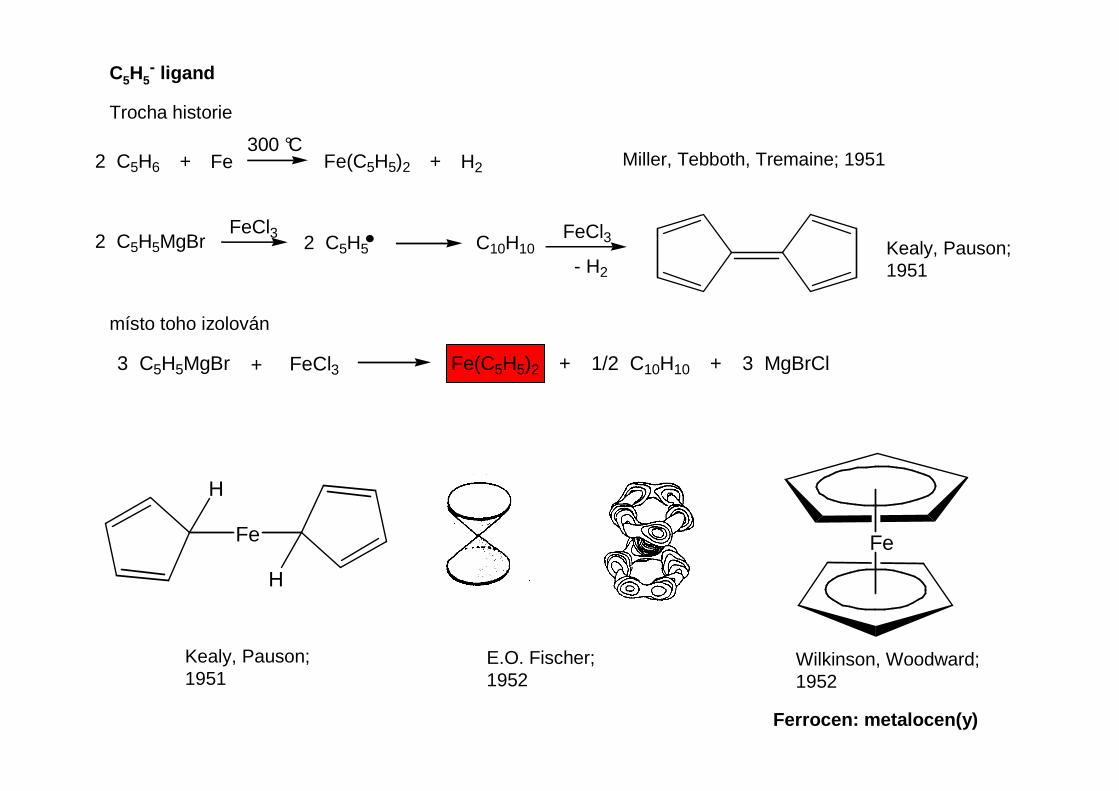
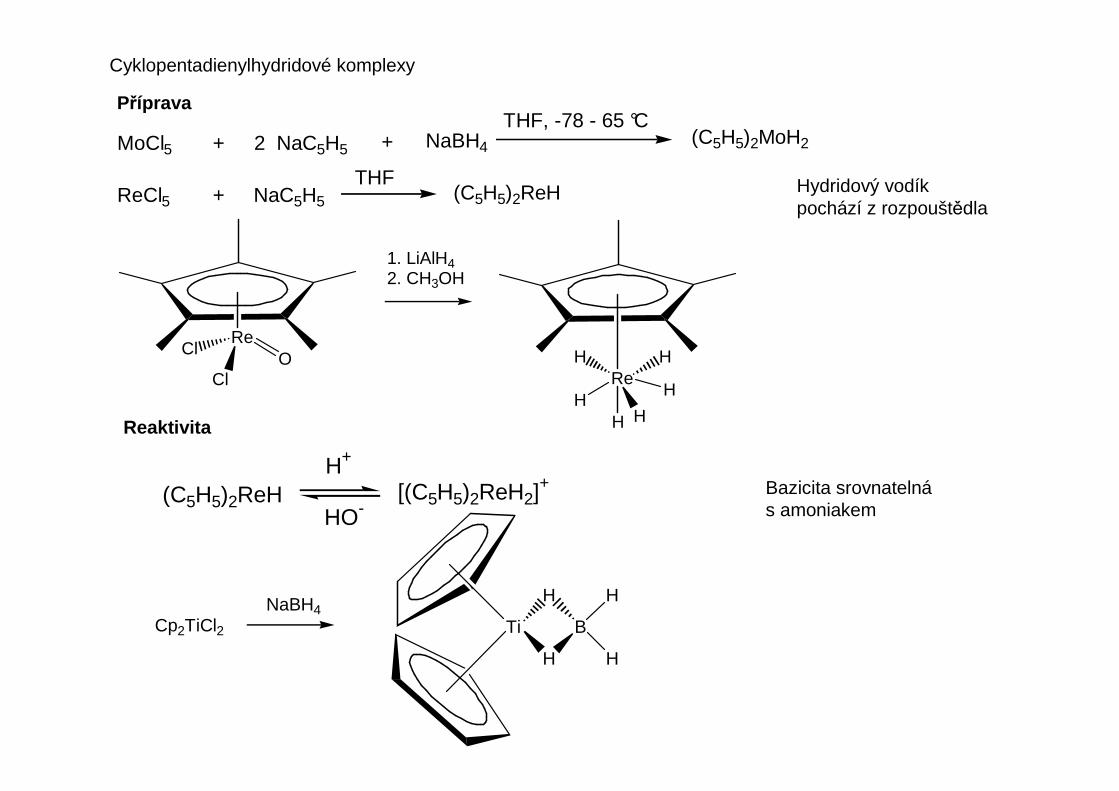
Prakticky důležitou sloučeninou je dicyklopentadienylhořčík (magnesocen)
EtMgBr CpMgBr220 °C, 10 Pa
MgCp2
500 °CMg + 2
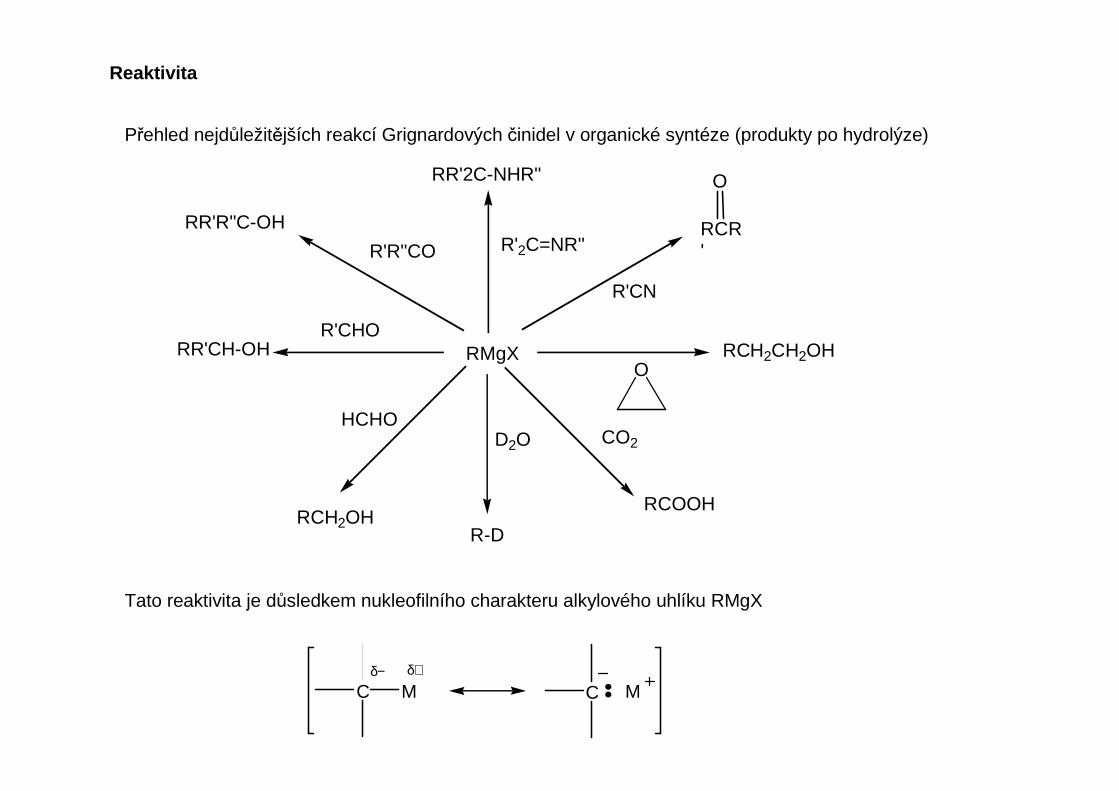
Přehled nejdůležitějších reakcí Grignardových činidel v organické syntéze (produkty po hydrolýze)
RMgX
HCHO
R'CHO
R'R''CO R'2C=NR''
R'CN
O
CO2D2O
RR'2C-NHR''
RCR'
O
RCH2CH2OH
RCOOH
R-DRCH2OH
RR'CH-OH
RR'R''C-OH
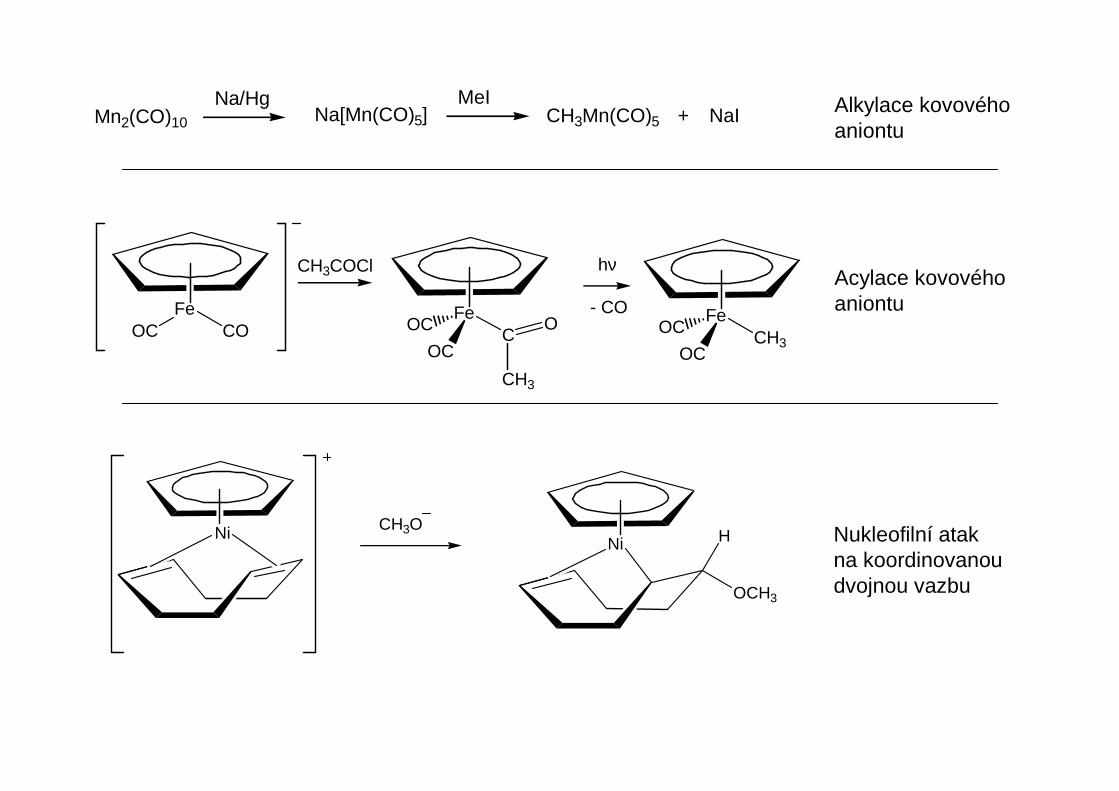
Tato reaktivita je důsledkem nukleofilního charakteru alkylového uhlíku RMgX
C M Cδ+δ−
M
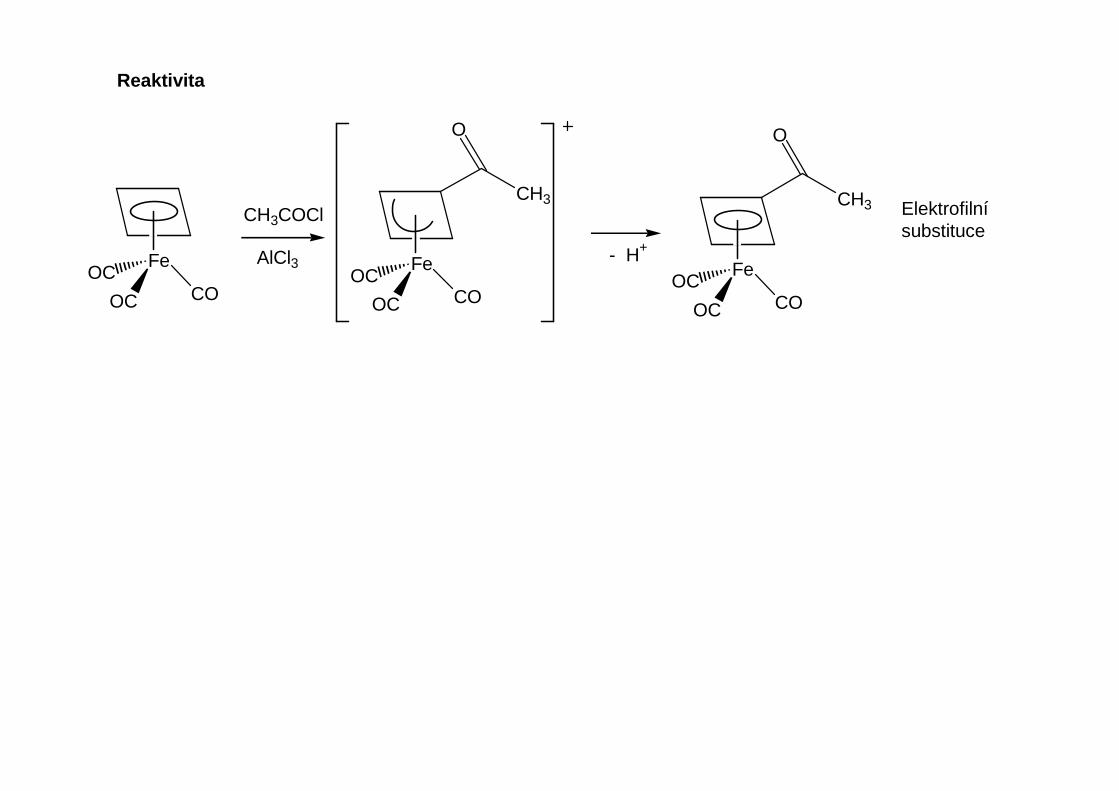
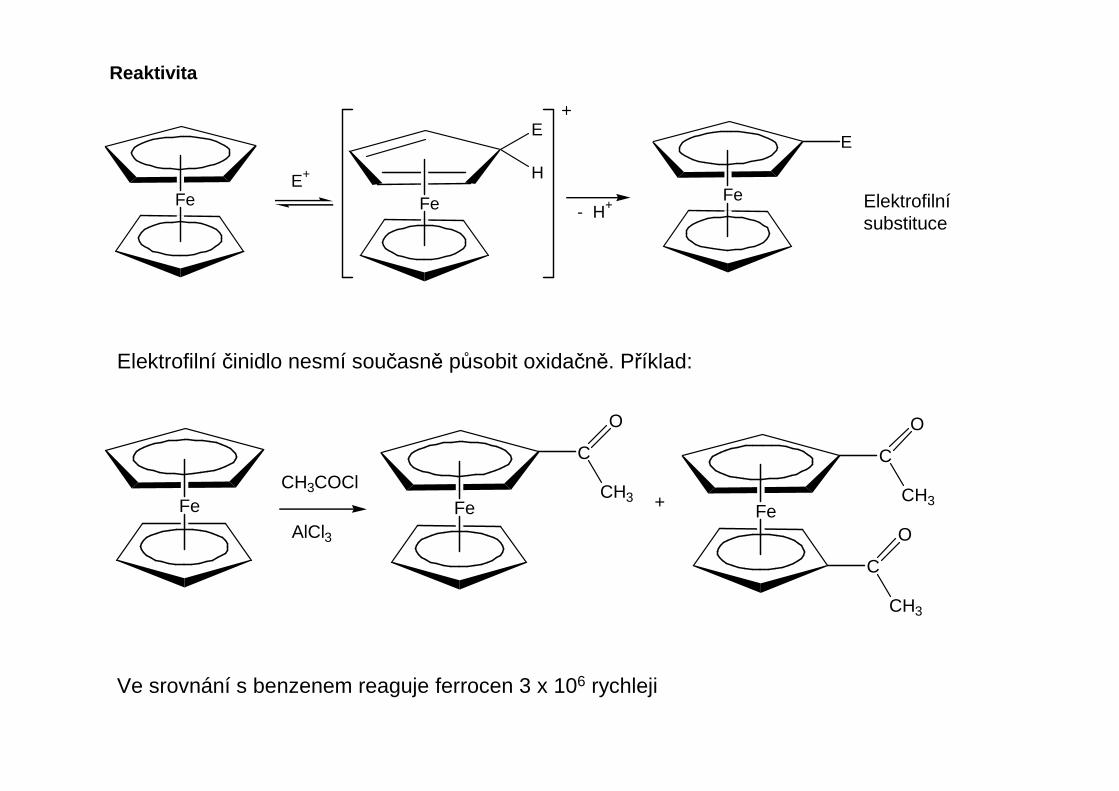
Reaktivita
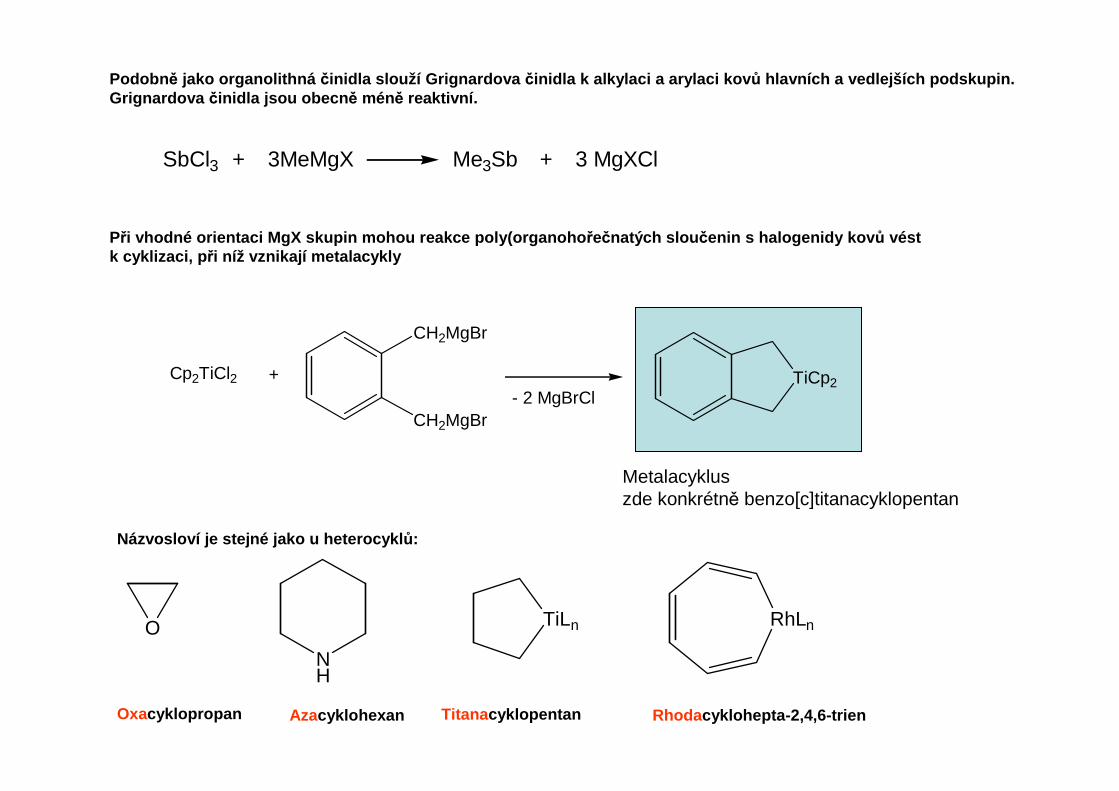
Podobn ě jako organolithná činidla slouží Grignardova činidla k alkylaci a arylaci kov ů hlavních a vedlejších podskupin.Grignardova činidla jsou obecn ě méně reaktivní.
SbCl3 + 3MeMgX Me3Sb + 3 MgXCl
Při vhodné orientaci MgX skupin mohou reakce poly(orga noho řečnatých slou čenin s halogenidy kov ů véstk cyklizaci, p ři níž vznikají metalacykly
Cp2TiCl2 +
CH2MgBr
CH2MgBr- 2 MgBrCl
TiCp2
Metalacykluszde konkrétně benzo[c]titanacyklopentan
E. Frankland, student R.W. Bunsena, se v roce1849 pokoušel připravit ethylový radikál reakcíethyljodidu se zinkem. Získal pyroforickou kapalinudiethylzinek (Et2Zn) a později řadu podobnýchorganokovových sloučenin. Je autorem termínuorganokovové slou čeniny .Ve Franklandově době se jako inertního plynupoužívalo vodíku ve skleněných aparaturách, který v kombinaci s pyroforickýmiprodukty syntéz představoval nebezpečí, kteréby dnes jen málokdo byl ochoten podstoupit.V jedné práci popisuje Frankland, jak při destilacikapalného organokovu došlo k jeho drobnému únikua vyšlehnutí plamene vysokého přes tři metryz aparatury.
Organozinečnaté sloučeniny
Příprava
Zn(Cu) + EtZnI +
Zn + R2Zn +R2Hg Hg
ZnCl2 + R2Zn + 2 LiCl (MgXCl)2 RLi (RMgX)
EtI Et2Zn ZnI2slitina
3 Zn(OAc)2 + 3 R2Zn + 2 Al(OAc)32 R3Al
Reakce
Organozinečnaté sloučeniny podléhají analogickým reakcím jako organohořečnaté a organolithné, jsou však méně reaktivní. Organozinečnatá činidla tolerují mnoho funkčních skupin.
Organokademnaté sloučeniny jsou podobné svým analogům z nižších řad periodického systému, dochází však k dalšímu snížení reaktivity.
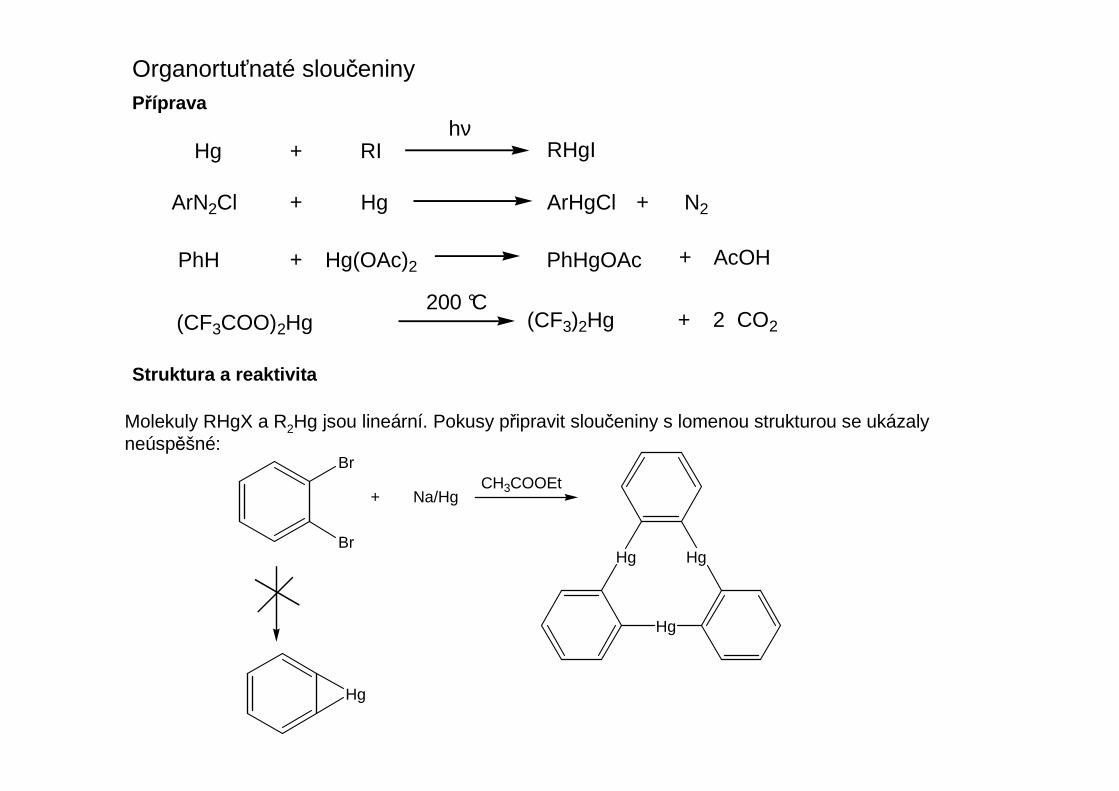
Organortuťnaté sloučeninyPříprava
Hg + RHgIRIhν
ArN2Cl + Hg ArHgCl + N2
PhH + Hg(OAc)2 PhHgOAc + AcOH
(CF3COO)2Hg (CF3)2Hg + 2 CO2200 °C
Struktura a reaktivita
Molekuly RHgX a R2Hg jsou lineární. Pokusy připravit sloučeniny s lomenou strukturou se ukázalyneúspěšné:
Br
Br
+ Na/HgCH3COOEt
Hg
Hg Hg
Hg
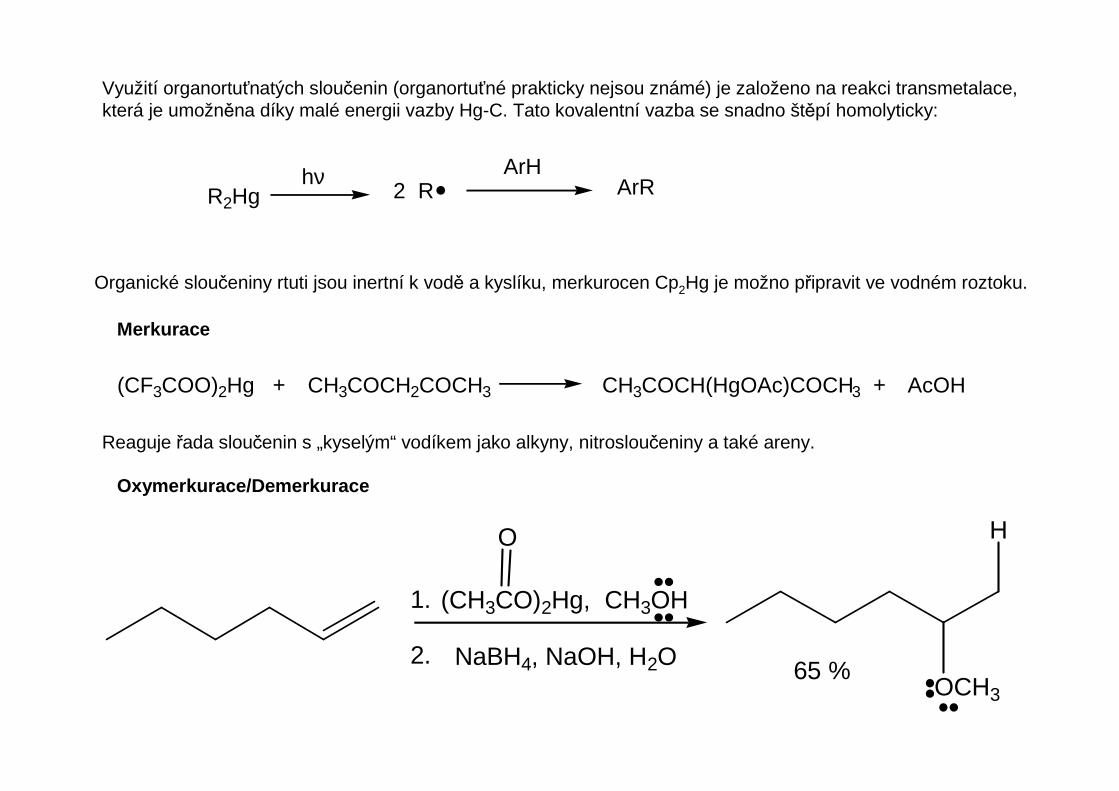
Využití organortuťnatých sloučenin (organortuťné prakticky nejsou známé) je založeno na reakci transmetalace,která je umožněna díky malé energii vazby Hg-C. Tato kovalentní vazba se snadno štěpí homolyticky:
Organické sloučeniny rtuti jsou inertní k vodě a kyslíku, merkurocen Cp2Hg je možno připravit ve vodném roztoku.
Reaguje řada sloučenin s „kyselým“ vodíkem jako alkyny, nitrosloučeniny a také areny.
Oxymerkurace/Demerkurace
R2Hghν
2 RArH
ArR
H
OCH3
(CH3CO)2Hg,
O
CH3OH1.
NaBH4, NaOH, H2O2.65 %
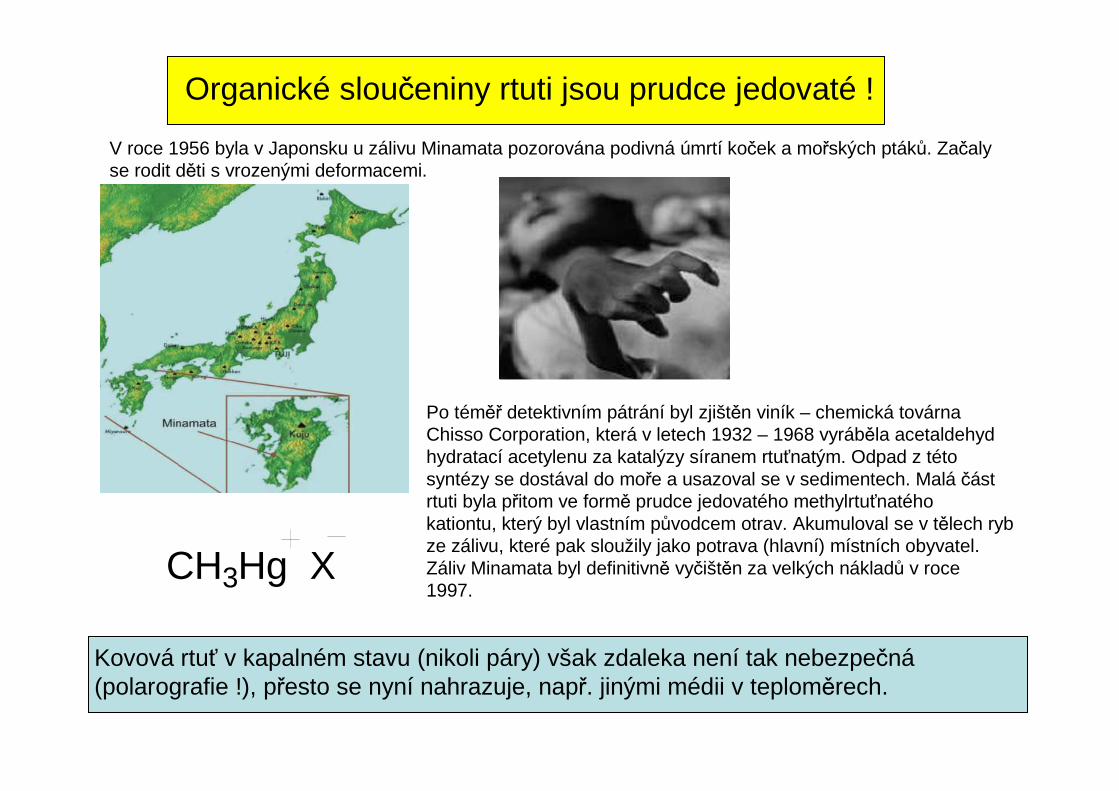
Organické sloučeniny rtuti jsou prudce jedovaté !
Kovová rtuť v kapalném stavu (nikoli páry) však zdaleka není tak nebezpečná(polarografie !), přesto se nyní nahrazuje, např. jinými médii v teploměrech.
V roce 1956 byla v Japonsku u zálivu Minamata pozorována podivná úmrtí koček a mořských ptáků. Začalyse rodit děti s vrozenými deformacemi.
Po téměř detektivním pátrání byl zjištěn viník – chemická továrnaChisso Corporation, která v letech 1932 – 1968 vyráběla acetaldehydhydratací acetylenu za katalýzy síranem rtuťnatým. Odpad z tétosyntézy se dostával do moře a usazoval se v sedimentech. Malá část rtuti byla přitom ve formě prudce jedovatého methylrtuťnatéhokationtu, který byl vlastním původcem otrav. Akumuloval se v tělech rybze zálivu, které pak sloužily jako potrava (hlavní) místních obyvatel.Záliv Minamata byl definitivně vyčištěn za velkých nákladů v roce1997.
CH3Hg X
Skupina 13 (3. hlavní podskupina): B, Al, ostatní kovy
Skupina 14 (4. hlavní podskupina): Si, Sn, Pb
Organické sloučeniny boru
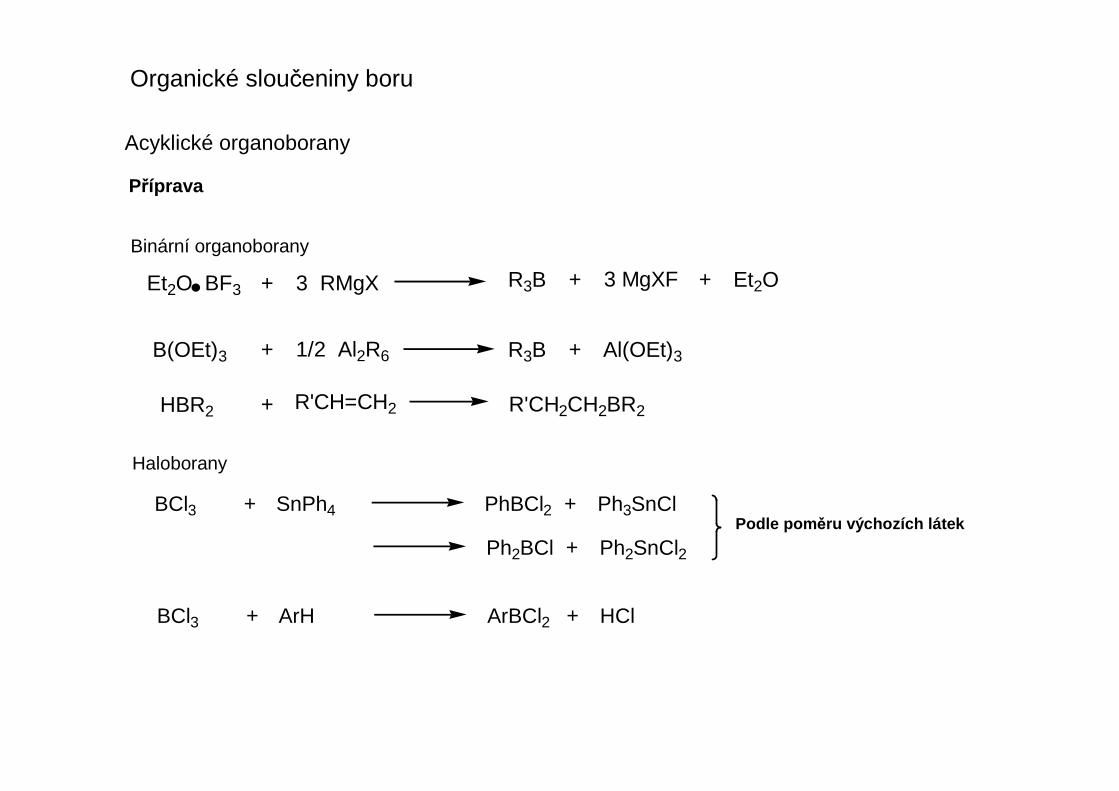
Acyklické organoborany
Příprava
Et2O BF3 + 3 RMgX R3B + 3 MgXF + Et2O
B(OEt)3 + 1/2 Al2R6 R3B + Al(OEt)3
HBR2 + R'CH2CH2BR2R'CH=CH2
BCl3 + SnPh4 PhBCl2 + Ph3SnCl
Ph2BCl + Ph2SnCl2
BCl3 + ArH ArBCl2 + HCl
Podle pom ěru výchozích látek
Binární organoborany
Haloborany
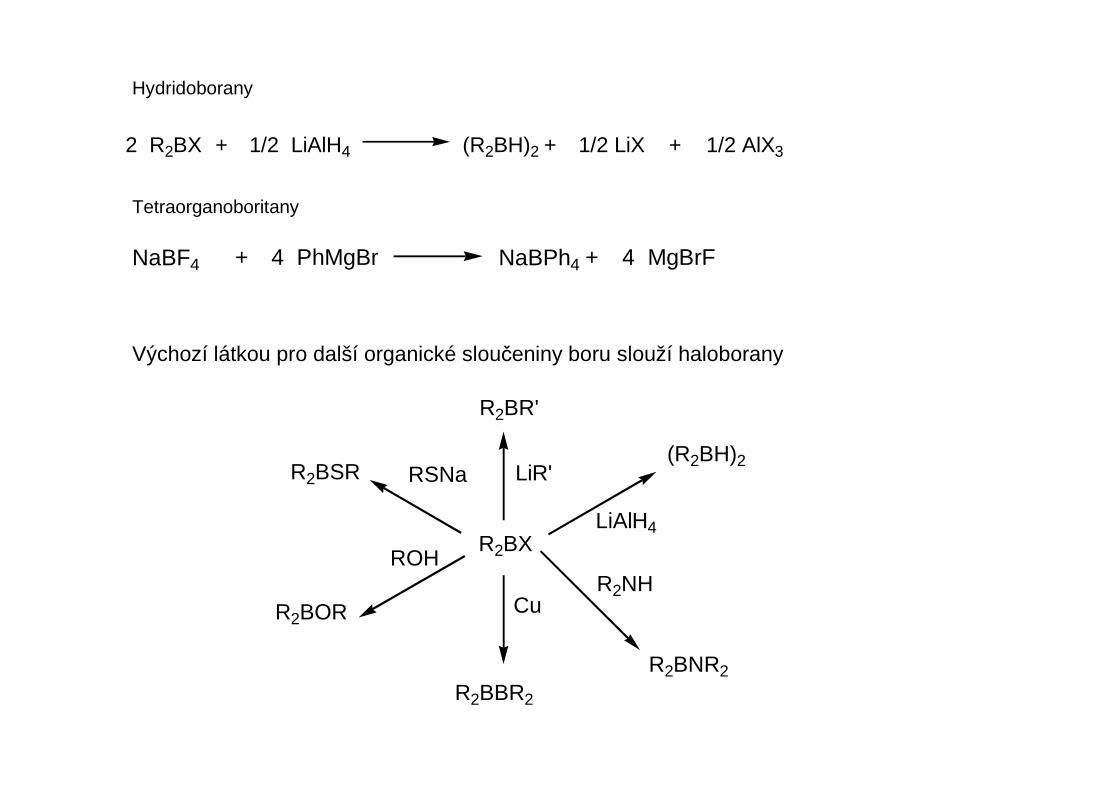
Hydridoborany
2 R2BX + 1/2 LiAlH4 (R2BH)2 + 1/2 LiX + 1/2 AlX3
Tetraorganoboritany
NaBF4 + 4 PhMgBr NaBPh4 + 4 MgBrF
R2BX
R2BR'
LiR'
LiAlH4
R2NHCu
ROH
RSNaR2BSR
R2BOR
(R2BH)2
R2BNR2
R2BBR2
Výchozí látkou pro další organické sloučeniny boru slouží haloborany
Struktura a reaktivita
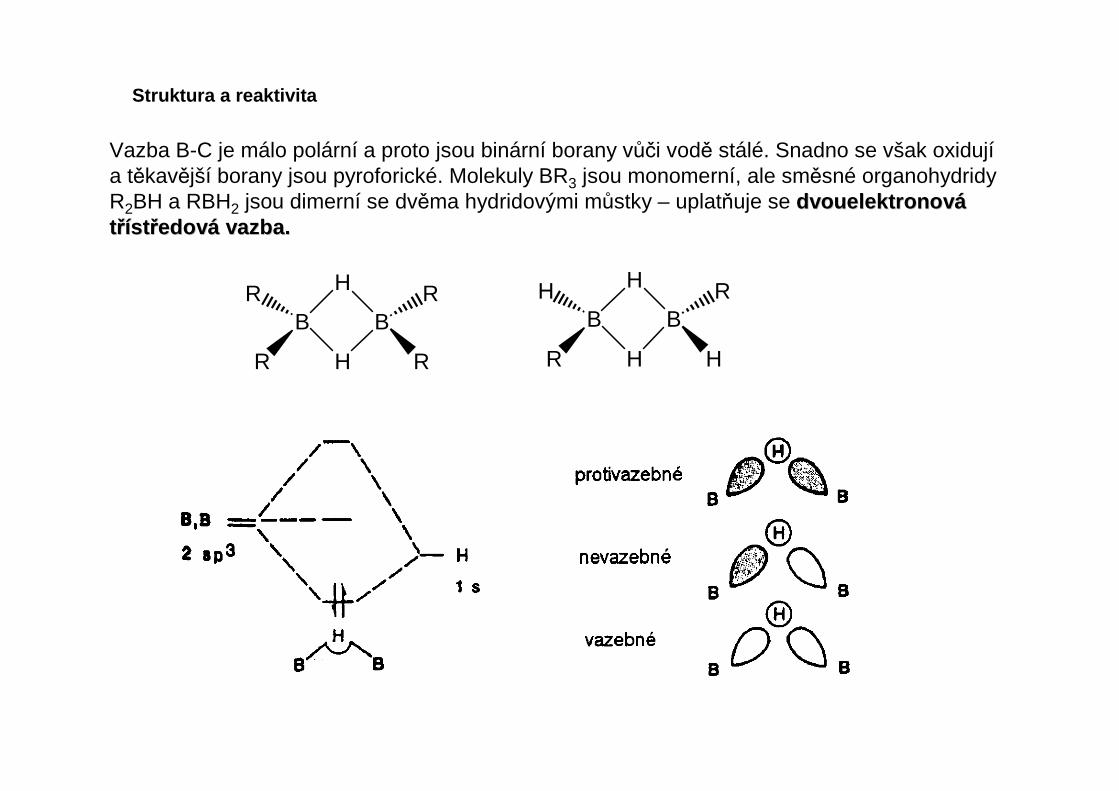
Vazba B-C je málo polární a proto jsou binární borany vůči vodě stálé. Snadno se však oxidujía těkavější borany jsou pyroforické. Molekuly BR3 jsou monomerní, ale směsné organohydridyR2BH a RBH2 jsou dimerní se dvěma hydridovými můstky – uplatňuje se dvouelektronovdvouelektronov ááttřříístst řředovedov áá vazba.vazba.
B
H
B
H
RR
RRB
H
B
H
HR
RH
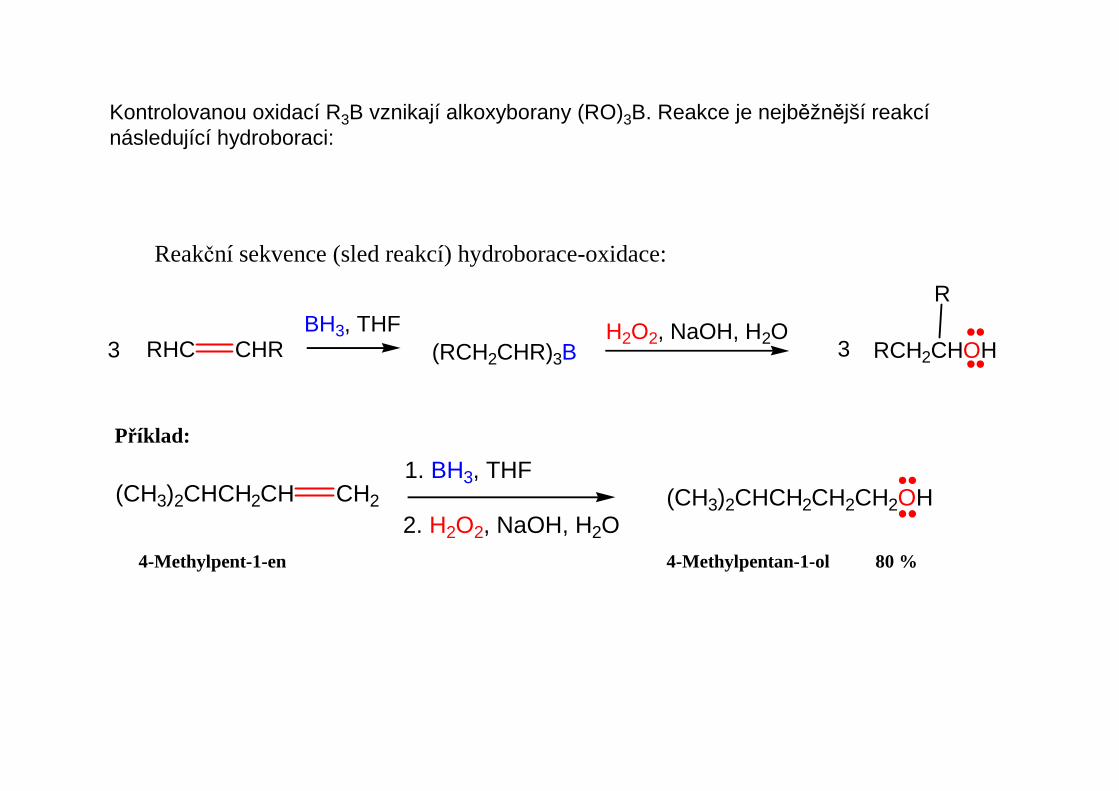
Kontrolovanou oxidací R3B vznikají alkoxyborany (RO)3B. Reakce je nejběžnější reakcínásledující hydroboraci:
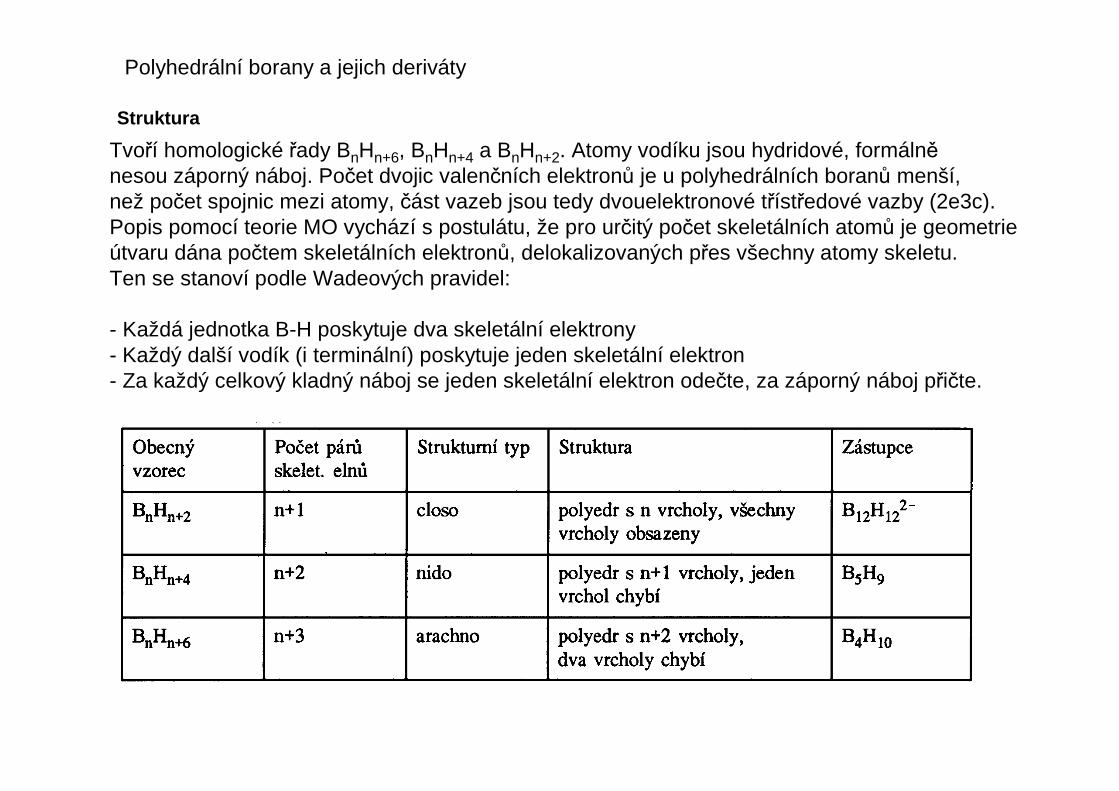
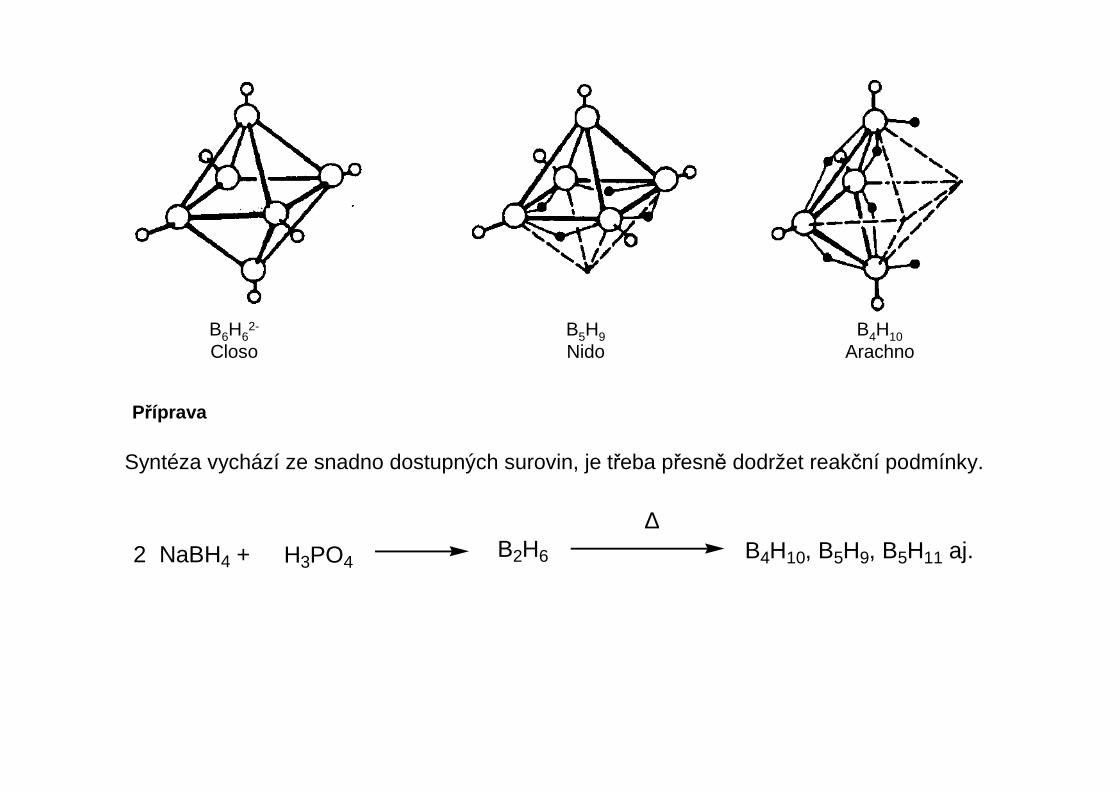
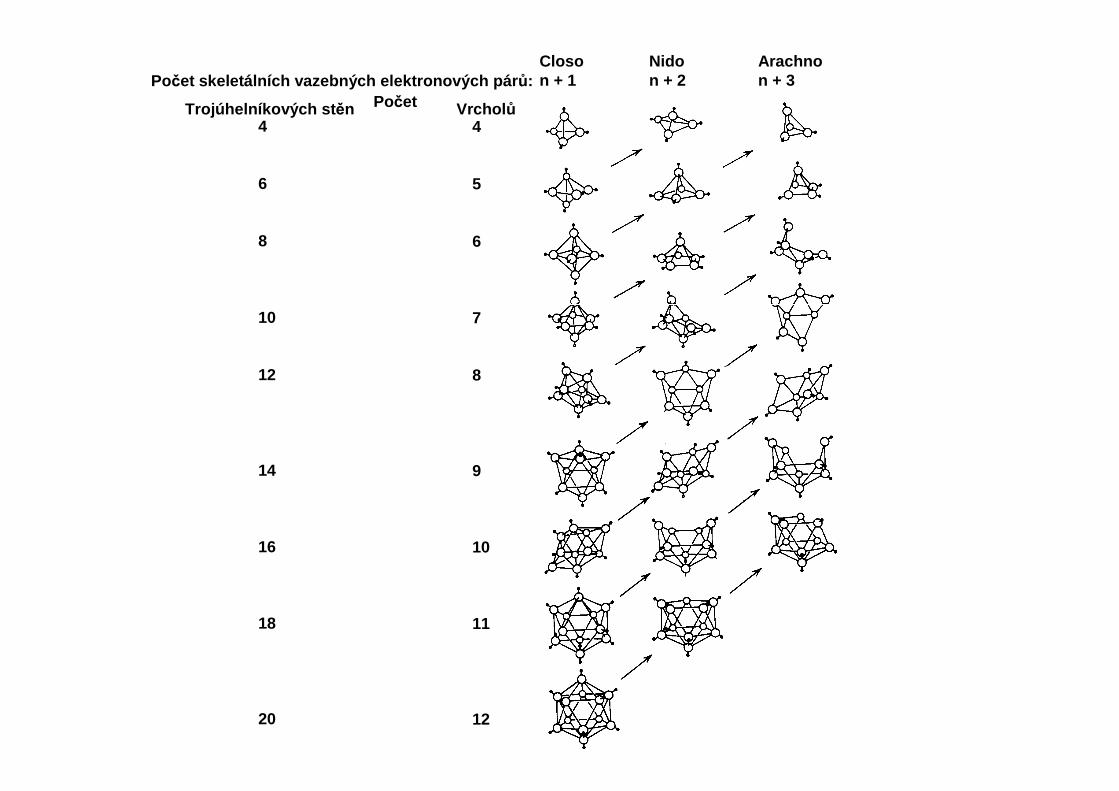
Tvoří homologické řady BnHn+6, BnHn+4 a BnHn+2. Atomy vodíku jsou hydridové, formálněnesou záporný náboj. Počet dvojic valenčních elektronů je u polyhedrálních boranů menší,než počet spojnic mezi atomy, část vazeb jsou tedy dvouelektronové třístředové vazby (2e3c).Popis pomocí teorie MO vychází s postulátu, že pro určitý počet skeletálních atomů je geometrieútvaru dána počtem skeletálních elektronů, delokalizovaných přes všechny atomy skeletu.Ten se stanoví podle Wadeových pravidel:
- Každá jednotka B-H poskytuje dva skeletální elektrony- Každý další vodík (i terminální) poskytuje jeden skeletální elektron- Za každý celkový kladný náboj se jeden skeletální elektron odečte, za záporný náboj přičte.
B6H62-
ClosoB5H9Nido
B4H10Arachno
Syntéza vychází ze snadno dostupných surovin, je třeba přesně dodržet reakční podmínky.
2 NaBH4 + H3PO4B2H6 B4H10, B5H9, B5H11 aj.
∆
Příprava
Closon + 1
Nidon + 2
Arachnon + 3Počet skeletálních vazebných elektronových pár ů:
PočetTrojúhelníkových st ěn Vrchol ů4
6
8
10
12
14
16
18
20
4
5
6
7
8
9
10
11
12
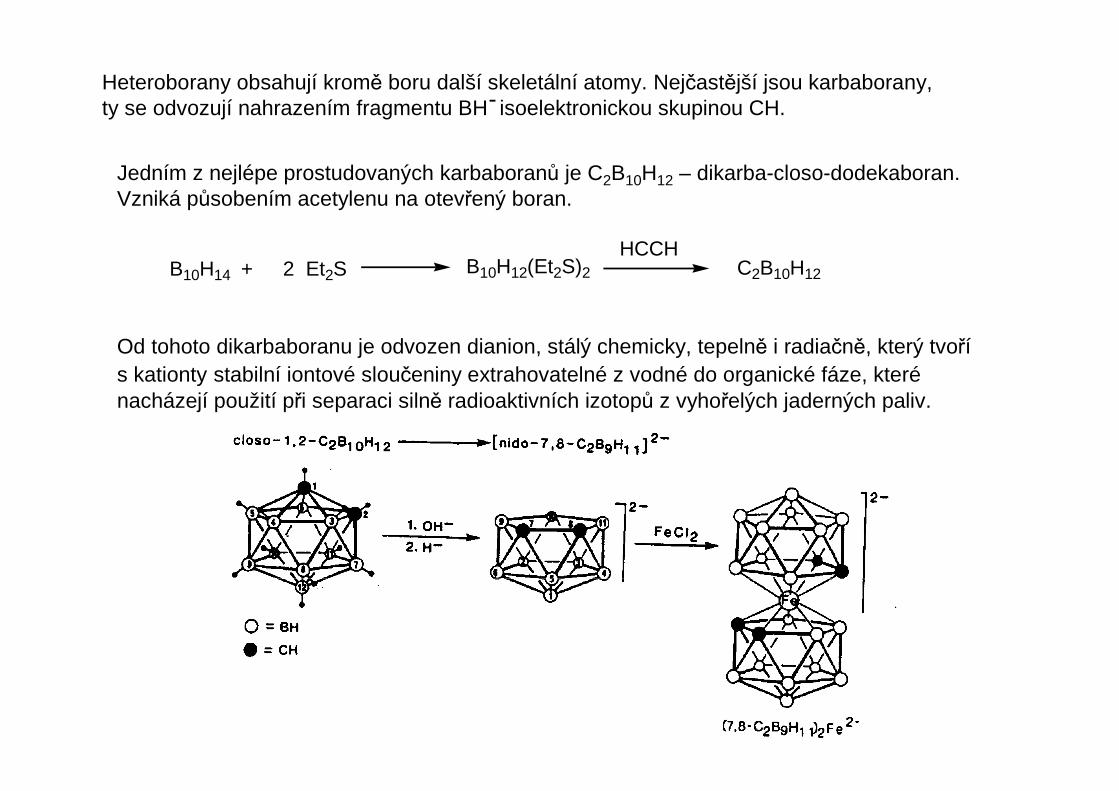
Heteroborany obsahují kromě boru další skeletální atomy. Nejčastější jsou karbaborany,ty se odvozují nahrazením fragmentu BH isoelektronickou skupinou CH.-
Jedním z nejlépe prostudovaných karbaboranů je C2B10H12 – dikarba-closo-dodekaboran.Vzniká působením acetylenu na otevřený boran.
B10H14 + 2 Et2S B10H12(Et2S)2 C2B10H12
HCCH
Od tohoto dikarbaboranu je odvozen dianion, stálý chemicky, tepelně i radiačně, který tvořís kationty stabilní iontové sloučeniny extrahovatelné z vodné do organické fáze, kterénacházejí použití při separaci silně radioaktivních izotopů z vyhořelých jaderných paliv.
Organické sloučeniny hliníku
Příprava
Vzhledem ke svým vlastnostem (samozápalné na vzduchu) se organické sloučeniny hliníkuv laboratoři málo připravují a využívá se především průmyslově vyráběných sloučenin.
Reakcí s 2-methylpropenem obdobnou k posledně uvedené se dospěje k rovnovážné reakci,kdy je možno získávat méně těkavý triisobutylderivát jeho odstraňováním z reakční směsi
Al(akt.) + 3/2 H2 + 3 CH2=CMe2
Al(CH2CHMe2)3
100 °C, 200 bar
HAl(CH2CHMe2)2 + 3 CH2=CMe2
140 °C, 20 mbar
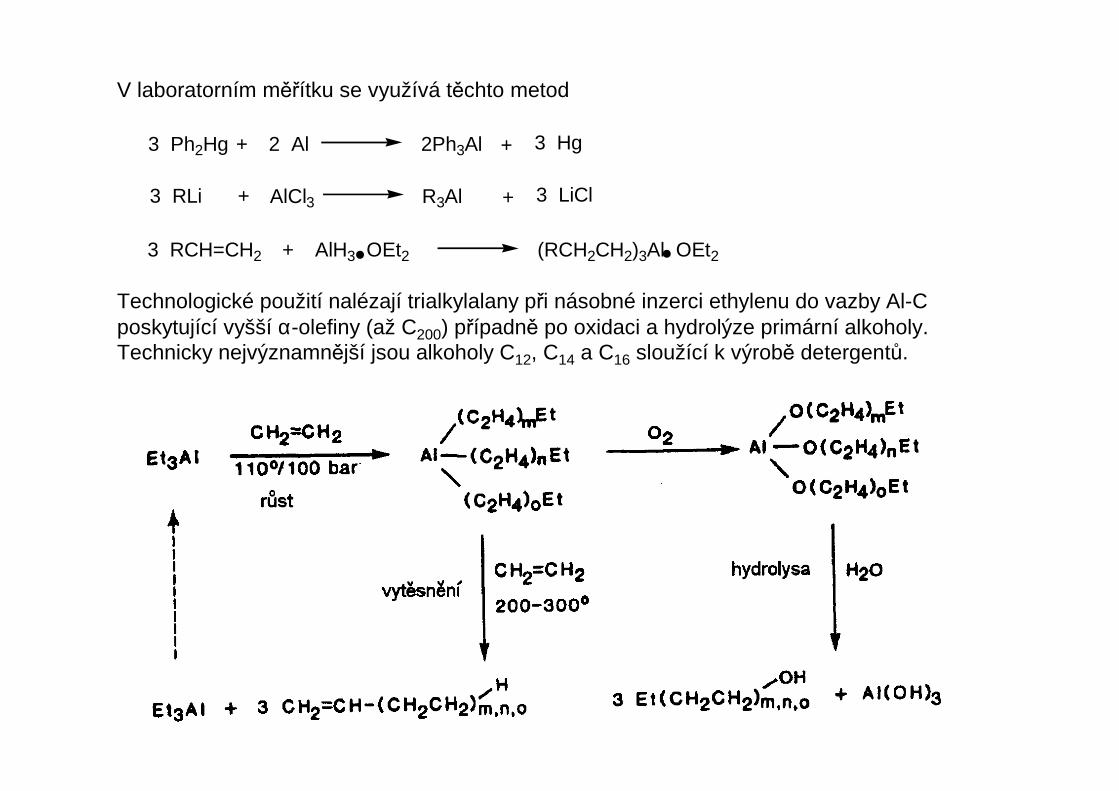
V laboratorním měřítku se využívá těchto metod
3 Ph2Hg + 2 Al 2Ph3Al + 3 Hg
3 RLi + + 3 LiClAlCl3 R3Al
3 RCH=CH2 + AlH3 OEt2 (RCH2CH2)3Al OEt2
Technologické použití nalézají trialkylalany při násobné inzerci ethylenu do vazby Al-Cposkytující vyšší α-olefiny (až C200) případně po oxidaci a hydrolýze primární alkoholy.Technicky nejvýznamnější jsou alkoholy C12, C14 a C16 sloužící k výrobě detergentů.
Struktura a reaktivita
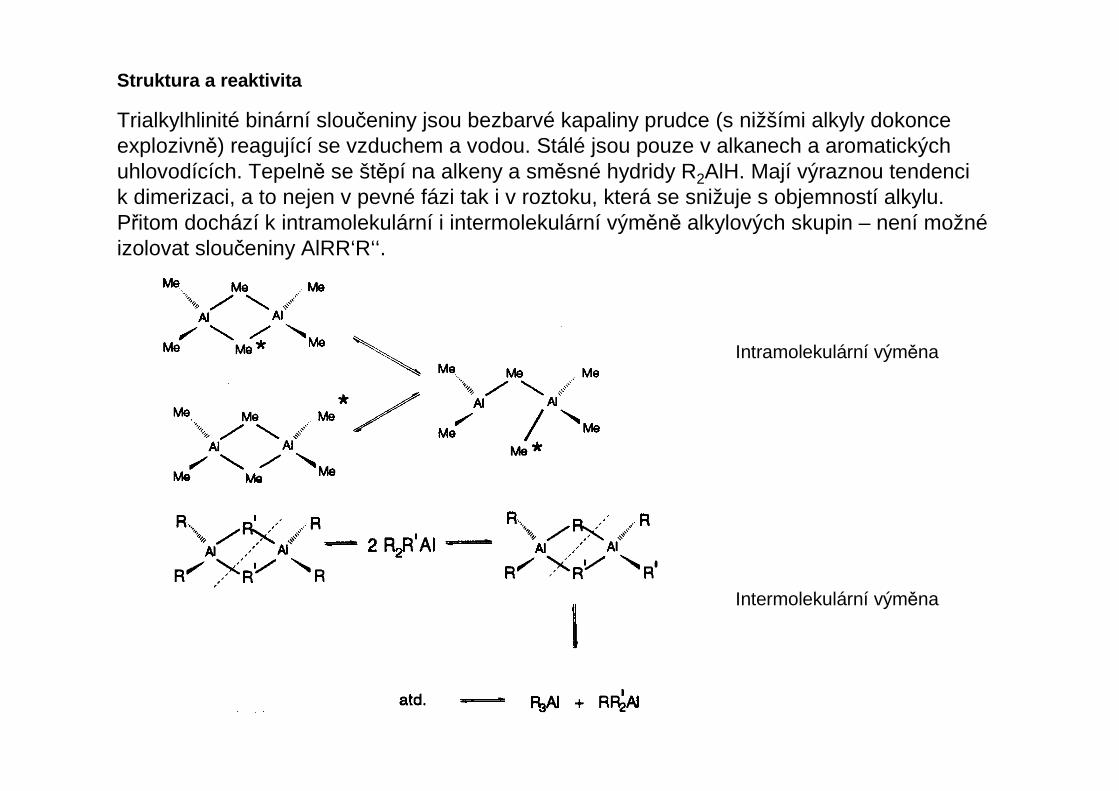
Trialkylhlinité binární sloučeniny jsou bezbarvé kapaliny prudce (s nižšími alkyly dokonceexplozivně) reagující se vzduchem a vodou. Stálé jsou pouze v alkanech a aromatických uhlovodících. Tepelně se štěpí na alkeny a směsné hydridy R2AlH. Mají výraznou tendencik dimerizaci, a to nejen v pevné fázi tak i v roztoku, která se snižuje s objemností alkylu.Přitom dochází k intramolekulární i intermolekulární výměně alkylových skupin – není možnéizolovat sloučeniny AlRR‘R‘‘.
Intramolekulární výměna
Intermolekulární výměna
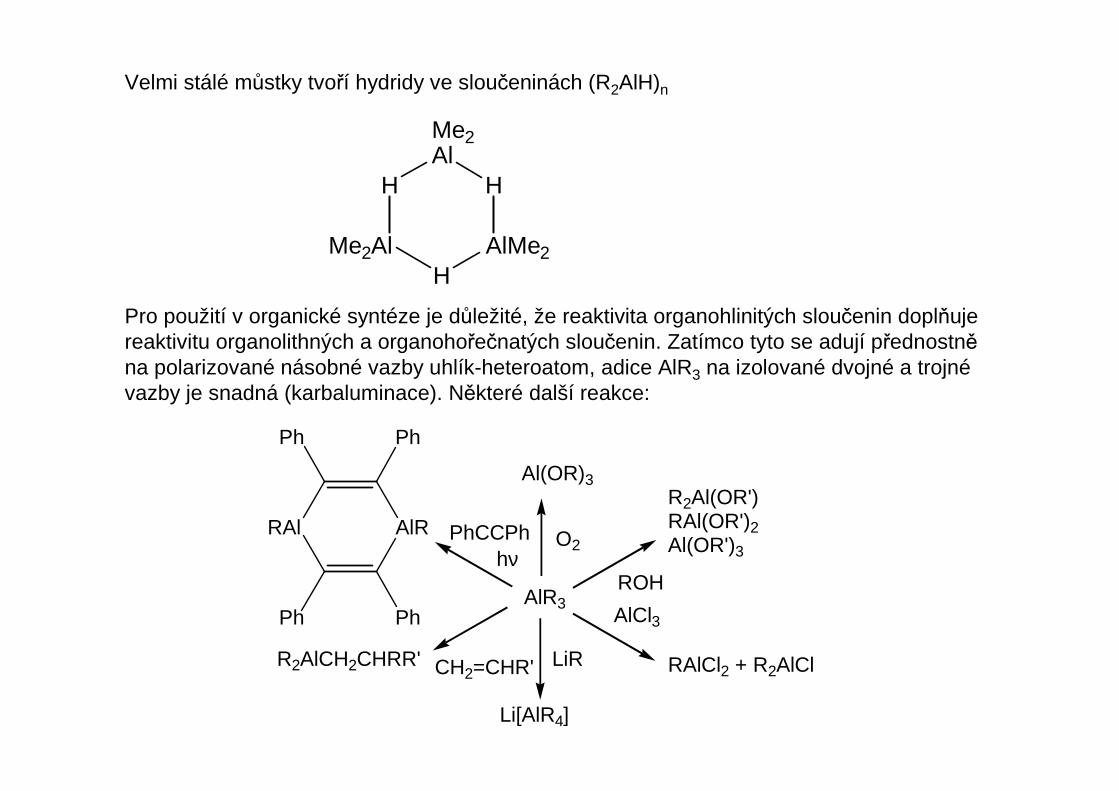
Velmi stálé můstky tvoří hydridy ve sloučeninách (R2AlH)n
Pro použití v organické syntéze je důležité, že reaktivita organohlinitých sloučenin doplňujereaktivitu organolithných a organohořečnatých sloučenin. Zatímco tyto se adují přednostněna polarizované násobné vazby uhlík-heteroatom, adice AlR3 na izolované dvojné a trojnévazby je snadná (karbaluminace). Některé další reakce:
H
Me2AlH
AlMe2
H
Me2Al
AlR3
O2
ROH
AlCl3
LiR
PhCCPh hν
CH2=CHR'
Al(OR)3R2Al(OR')RAl(OR')2Al(OR')3
RAlCl2 + R2AlCl
Li[AlR4]
R2AlCH2CHRR'
RAl AlR
PhPh
Ph Ph
Reakcí příbuznou hydroboraci je hydroaluminace, která je vysoce cis-stereoselektivní,regioselektivita je převážně antimarkovnikovovská, ale záleží na druhu R.
R2AlH + R'2C=CR'2 R'2C(AlR2)CHR'2
Výhodou proti hydroboraci je, že následná oxidace nevyžaduje peroxidy, nevýhodou menšítolerance funkčních skupin.
Organické sloučeniny gallia, india a thallia
Praktický význam těchto sloučenin ve srovnání se sloučeninami boru a hliníku je velmi malý.
I když jsou sloučeniny thallia značně jedovaté, našly použití v organické syntéze při některýchspeciálních reakcích. V chemii cyklopentadienylových komplexů je stále ceněným výchozímkomplexem CpTl, který se podobně jako Cp2Hg dá připravit ve vodném roztoku a je stálý:
Tl2SO4 + 2 C5H6 + 2 NaOH 2 CpTl + Na2SO4 + 2 H2O
Organické sloučeniny křemíku
Mnoho organosilanů se vyrábí průmyslově, např. takzvanou přímou syntézou (viz dále),v laboratoři se proto připravují jen ojediněle metathesou nebo hydrosilylací. Hydrosilylace je regiospecifická a antimarkovnikovovská, je katalyzována radikálově nebo komplexy kovů.
Příprava
SiCl4 + 4 RLi R4Si + 4 LiCl
R3SiCl + R'MgX R3SiR'+ MgXCl
2 R2SiCl2 + LiAlH4 2 R2SIH2 + LiCl + AlCl3
HSiCl3 + RCH=CH2 RCH2CH2SiCl3
Reaktivita
Homolytické štěpení Si-C vazeb probíhá až při značně vysokých teplotách, ani heterolytickéštěpení není snadné v důsledku nízké polarity Si-C vazby. Pokud mechanismus spočíváv elektrofilním ataku, je nutná katalýza silnou kyselinou.
Me4Si + HCl Me3SiCl + CH4
AlCl3
benzen
SiR3
H
HX
X = CF3COOH, RFSO3H
SiR3H
OH2
+ R3SiOH2
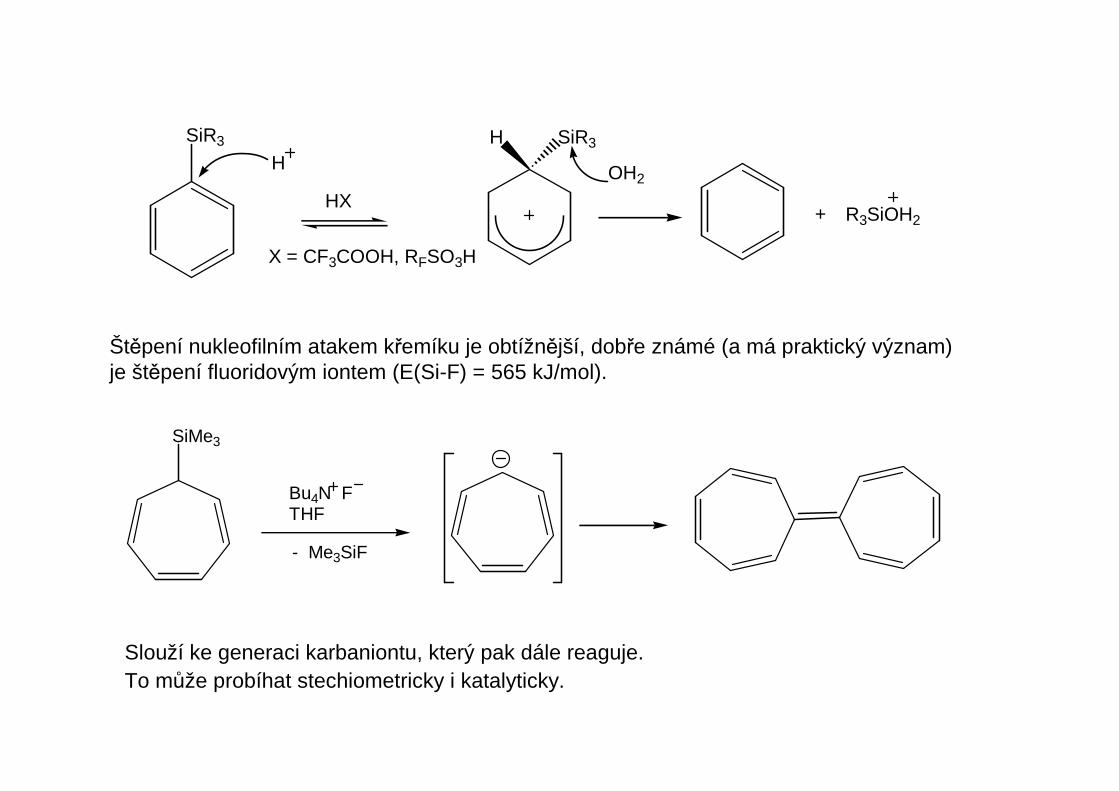
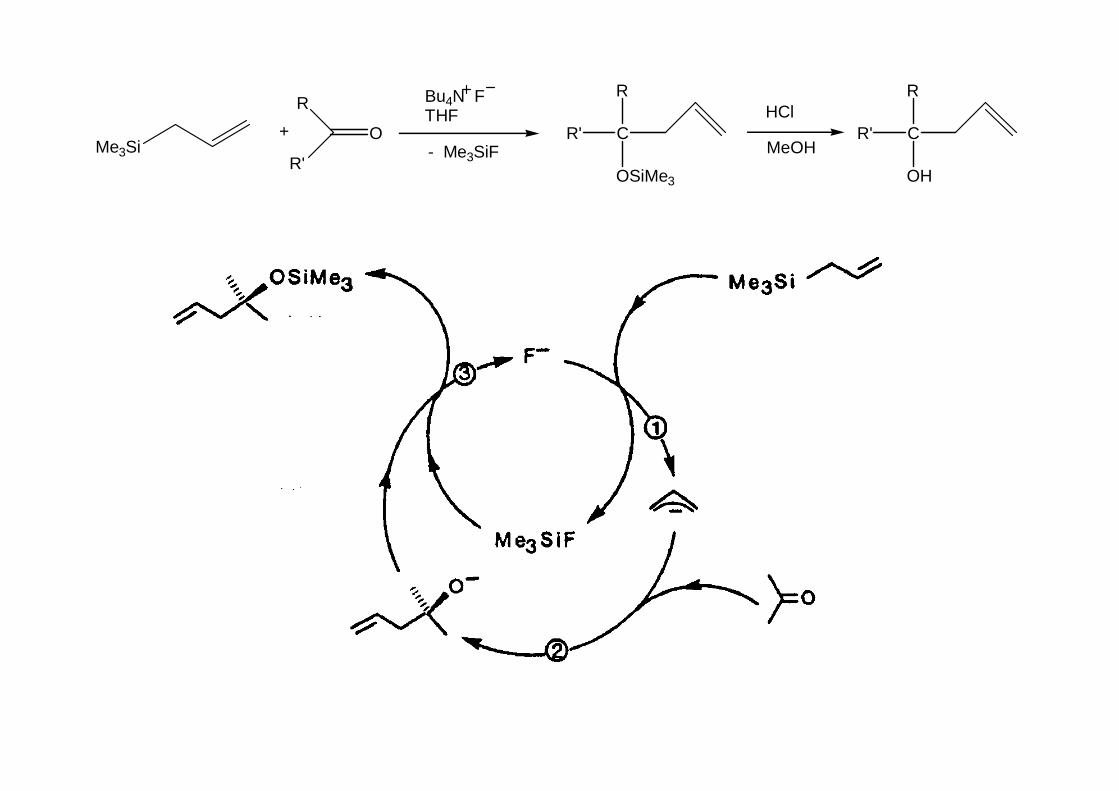
Štěpení nukleofilním atakem křemíku je obtížnější, dobře známé (a má praktický význam)je štěpení fluoridovým iontem (E(Si-F) = 565 kJ/mol).
SiMe3
Bu4N FTHF
- Me3SiF
Slouží ke generaci karbaniontu, který pak dále reaguje. To může probíhat stechiometricky i katalyticky.
Me3Si+
R
O
R'
Bu4N FTHF
- Me3SiFR' C
R
OSiMe3
HCl
MeOHR' C
R
OH
Vazby Si-C zůstávají díky své nízké polaritě při mnoha významných reakcích nedotčeny. Chemie organokřemičitých sloučenin se tak, i díky převažující čtyřvaznosti křemíku, podobáorganické chemii uhlíku.
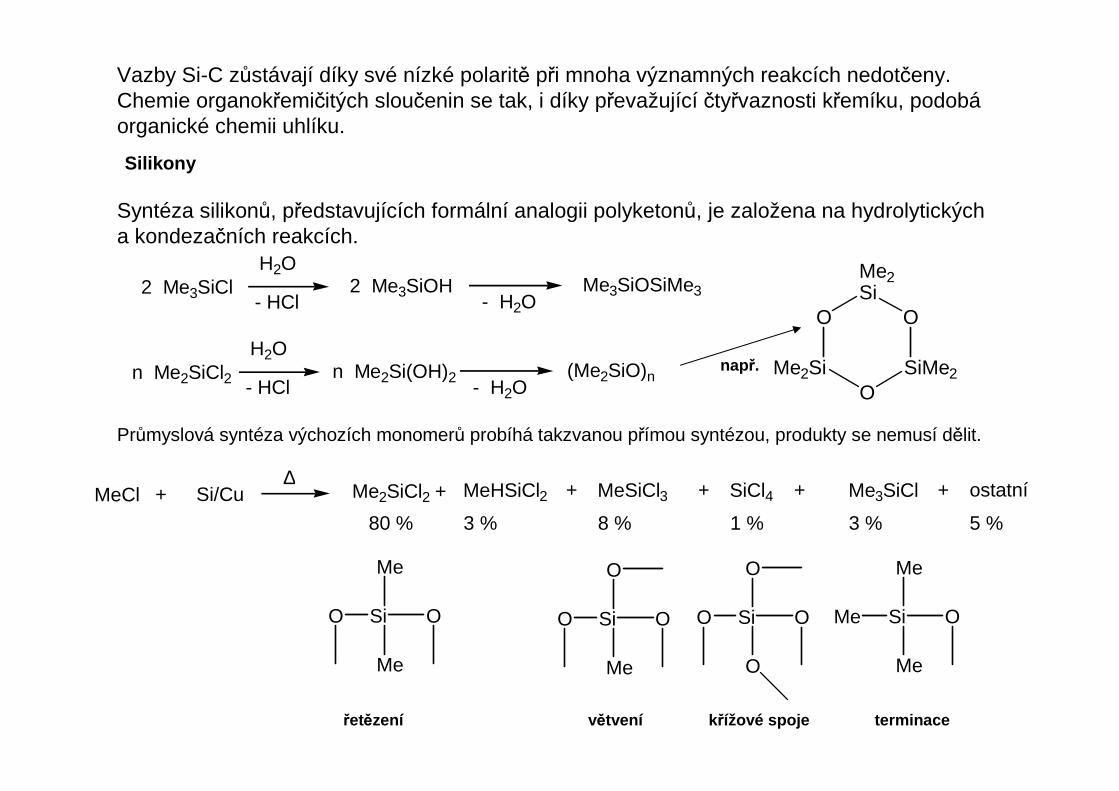
Silikony
Syntéza silikonů, představujících formální analogii polyketonů, je založena na hydrolytických a kondezačních reakcích.
2 Me3SiCl 2 Me3SiOH Me3SiOSiMe3
H2O
- HCl - H2O
n Me2SiCl2 n Me2Si(OH)2 (Me2SiO)n
H2O
- HCl - H2O
O
Me2SiO
SiMe2
O
Me2Si
např.
Průmyslová syntéza výchozích monomerů probíhá takzvanou přímou syntézou, produkty se nemusí dělit.
MeCl + Si/Cu∆
Me2SiCl2 + MeHSiCl2 + MeSiCl3 + SiCl4 +
Me
Si
Me
OO
O
Si
Me
OO
Me3SiCl + ostatní
O
Si
O
OO
Me
Si
Me
OMe
80 % 3 % 8 % 1 % 3 % 5 %
řetězení větvení k řížové spoje terminace
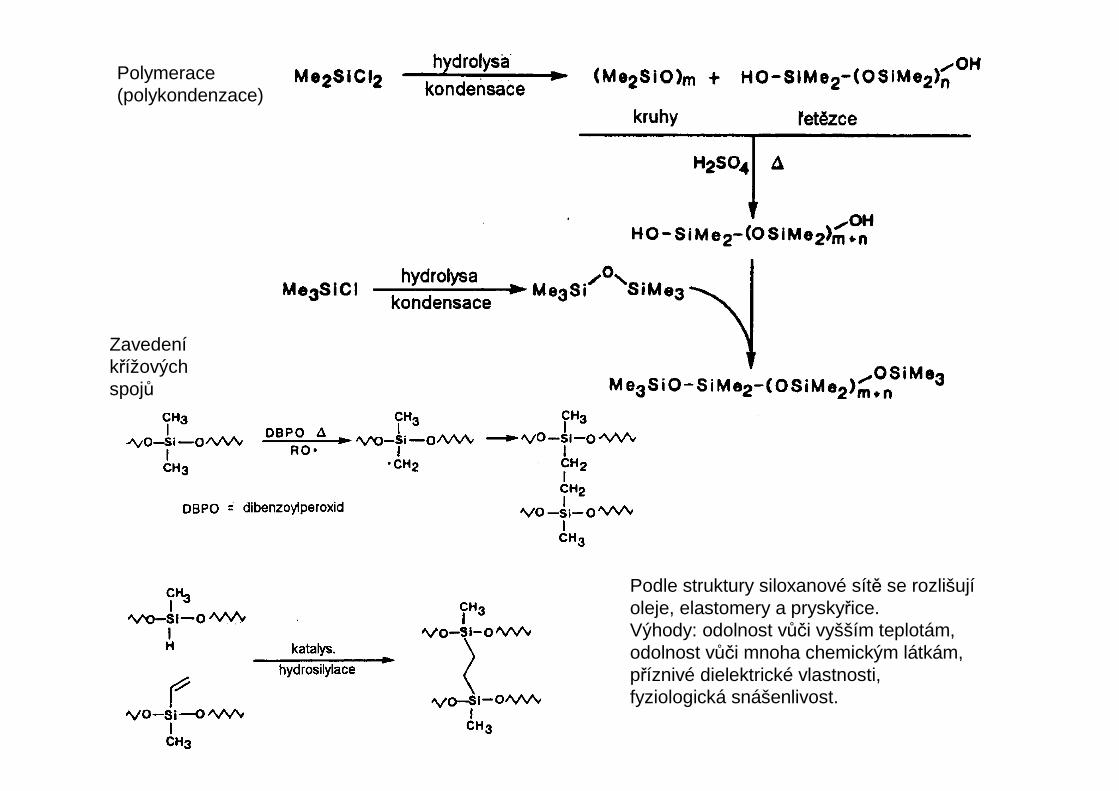
Polymerace (polykondenzace)
Zavedeníkřížových spojů
Podle struktury siloxanové sítě se rozlišujíoleje, elastomery a pryskyřice.Výhody: odolnost vůči vyšším teplotám,odolnost vůči mnoha chemickým látkám,příznivé dielektrické vlastnosti,fyziologická snášenlivost.
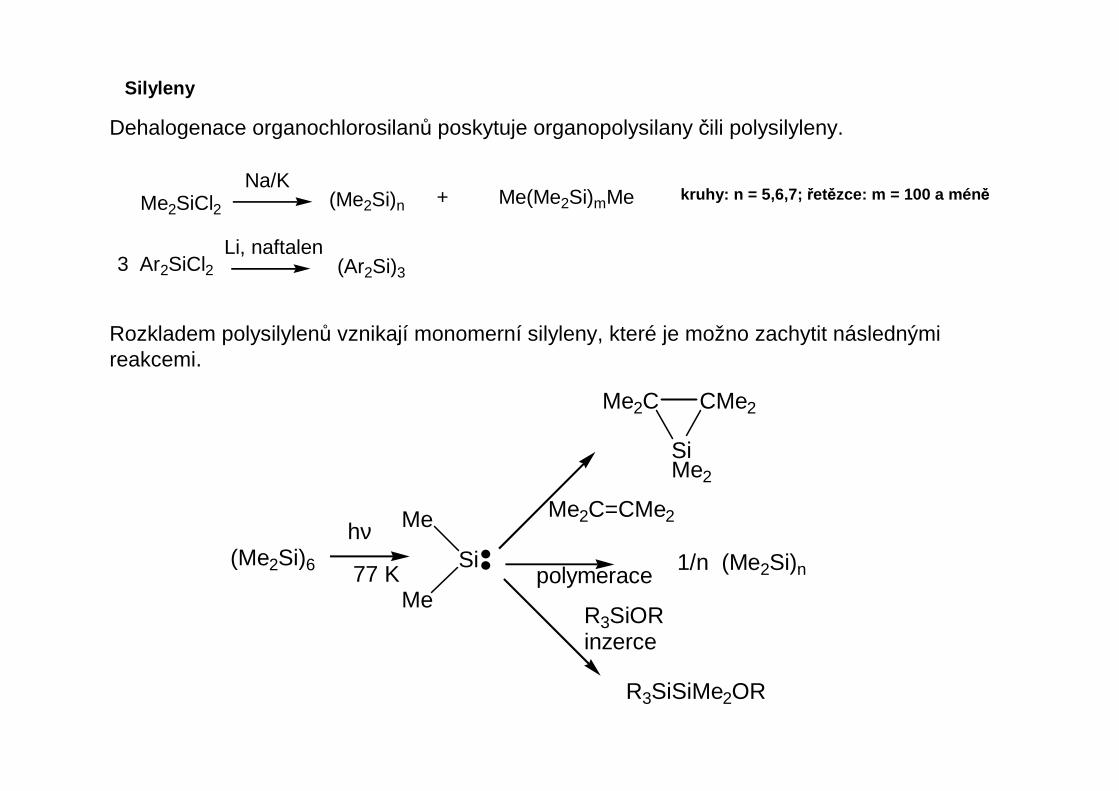
Silyleny
Dehalogenace organochlorosilanů poskytuje organopolysilany čili polysilyleny.
Me2SiCl2Na/K
(Me2Si)n + Me(Me2Si)mMe
3 Ar2SiCl2Li, naftalen
(Ar2Si)3
kruhy: n = 5,6,7; řetězce: m = 100 a mén ě
Rozkladem polysilylenů vznikají monomerní silyleny, které je možno zachytit následnými reakcemi.
(Me2Si)6
hν
77 K
Me
Si
Me
Me2C=CMe2
polymerace
R3SiORinzerce
Me2C
SiMe2
CMe2
1/n (Me2Si)n
R3SiSiMe2OR
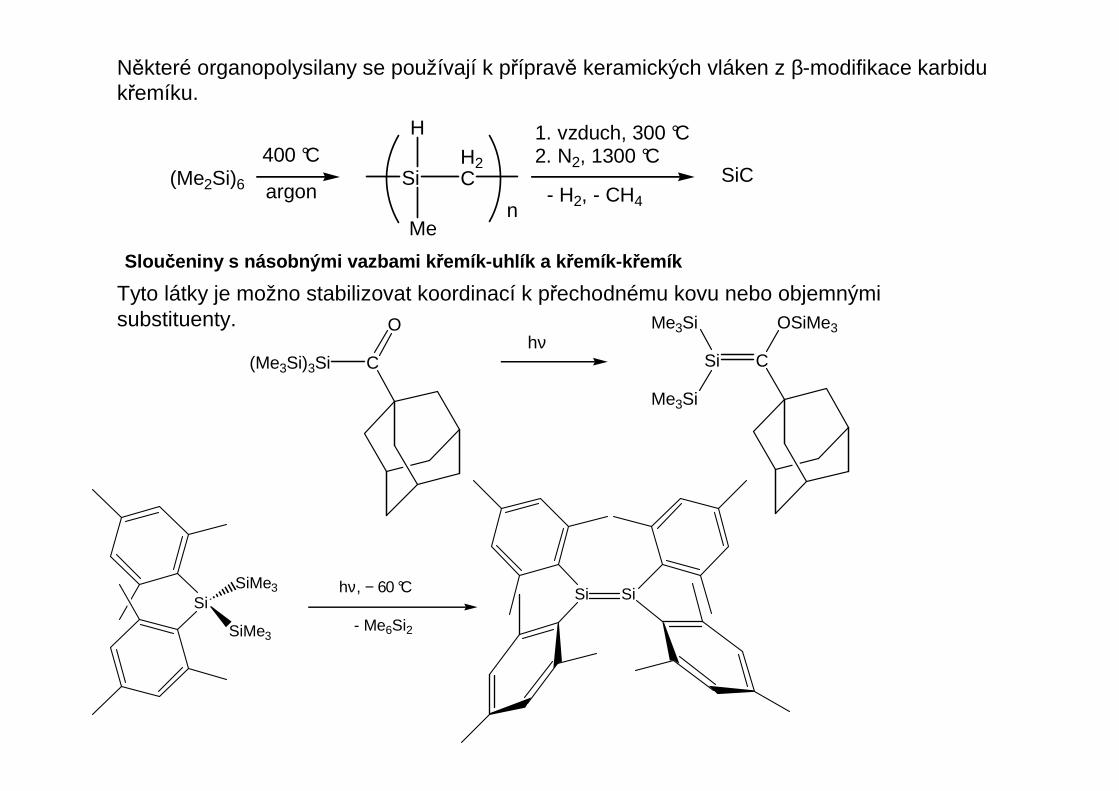
Některé organopolysilany se používají k přípravě keramických vláken z β-modifikace karbidukřemíku.
(Me2Si)6
400 °C
argonSi
H
Me
H2C
n
1. vzduch, 300 °C2. N2, 1300 °C
- H2, - CH4
SiC
Sloučeniny s násobnými vazbami k řemík-uhlík a k řemík-k řemík
Tyto látky je možno stabilizovat koordinací k přechodnému kovu nebo objemnými substituenty.
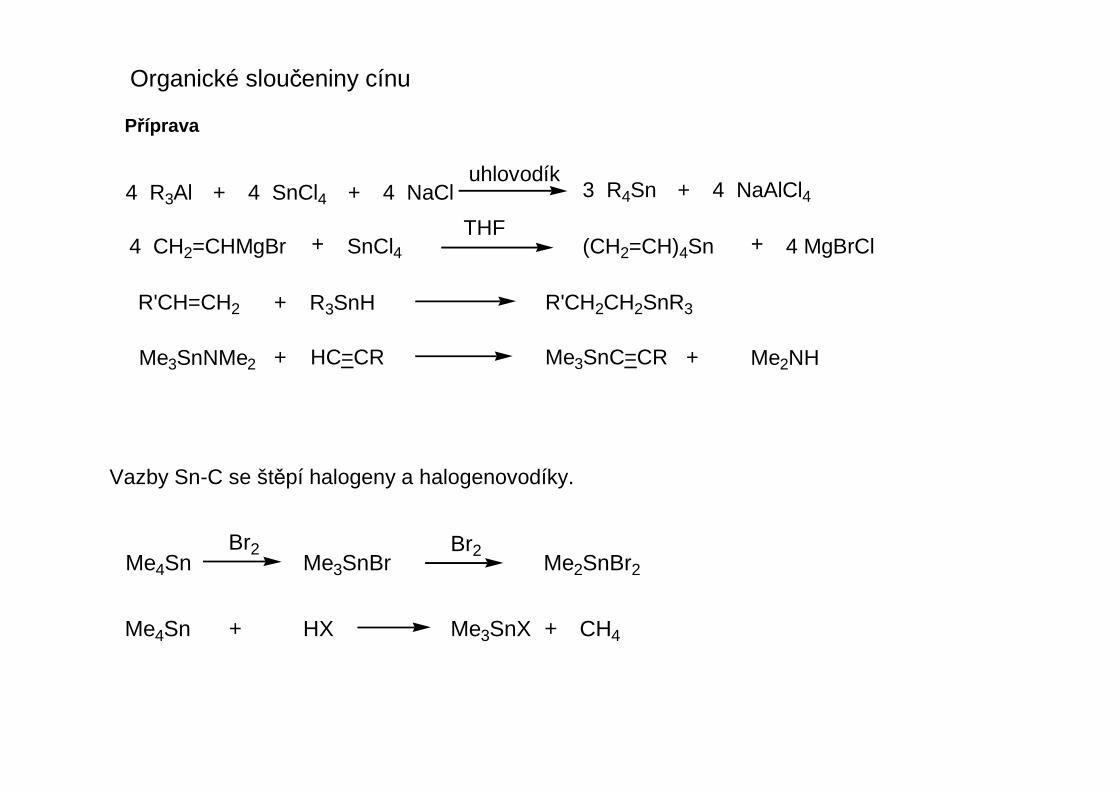
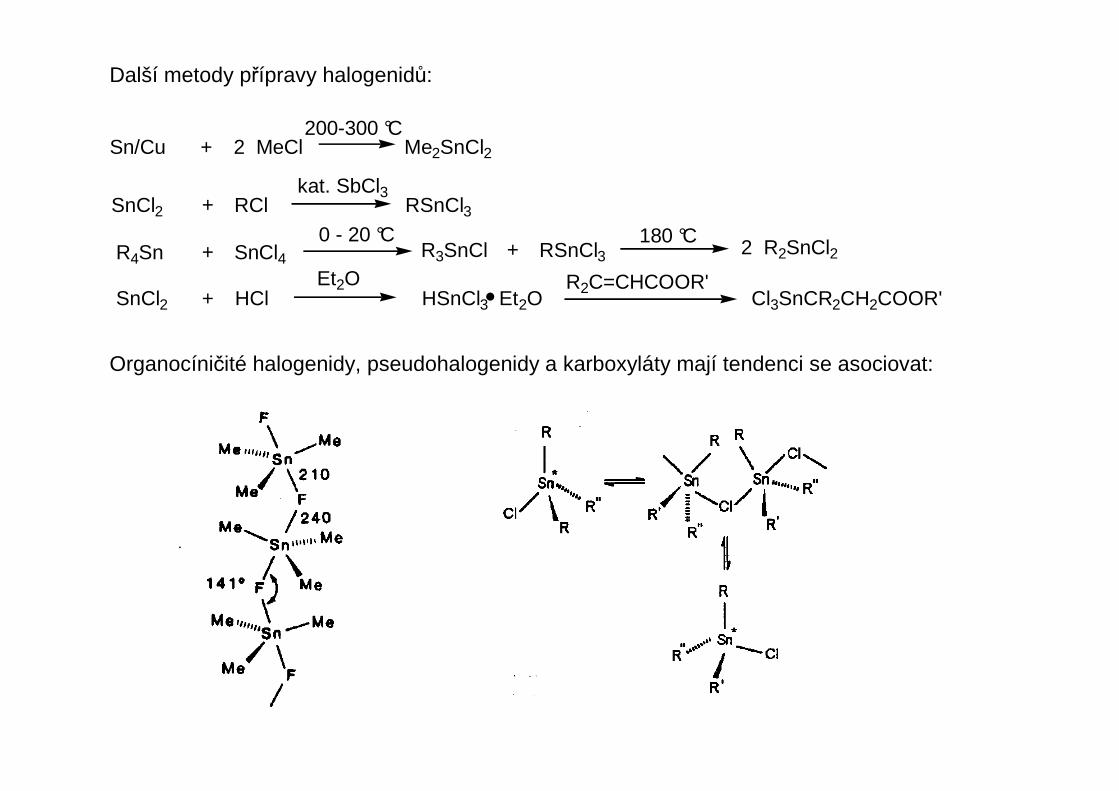
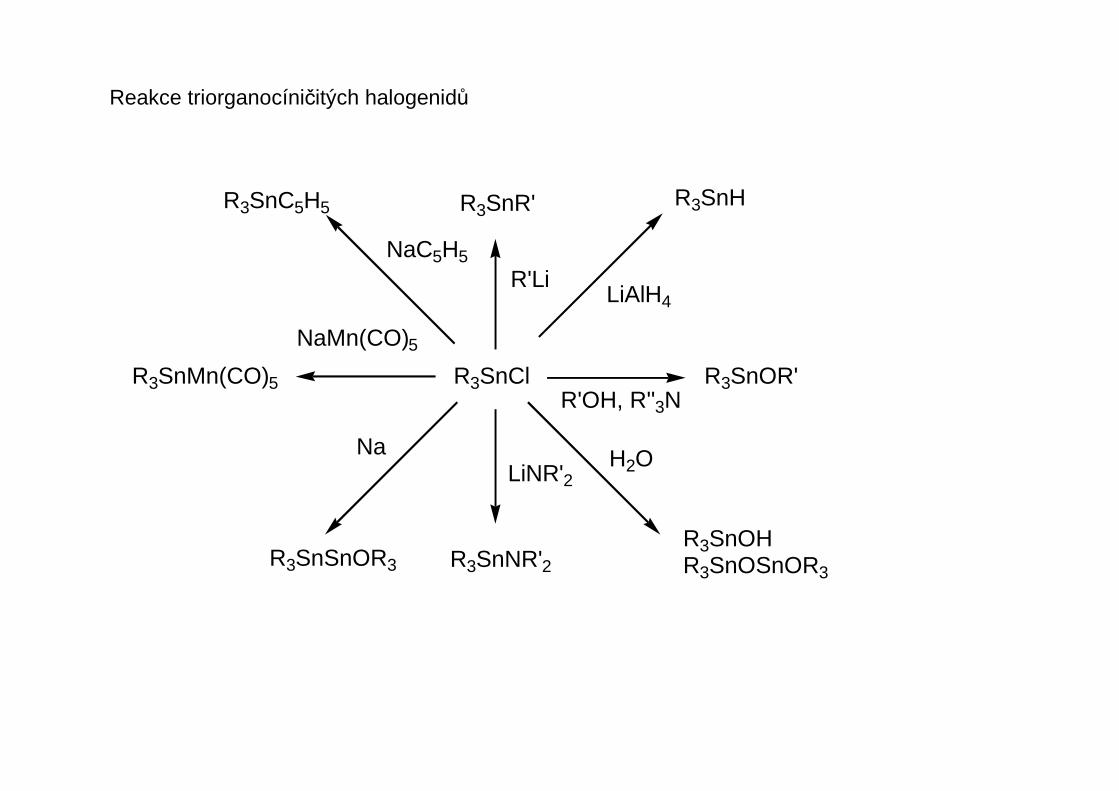
Organocíničité halogenidy, pseudohalogenidy a karboxyláty mají tendenci se asociovat:
R3SnCl
R3SnH
R3SnOR'
R3SnOHR3SnOSnOR3
R3SnSnOR3 R3SnNR'2
R3SnR'R3SnC5H5
R3SnMn(CO)5
NaMn(CO)5
NaC5H5
R'LiLiAlH4
R'OH, R''3N
H2OLiNR'2Na
Reakce triorganocíničitých halogenidů
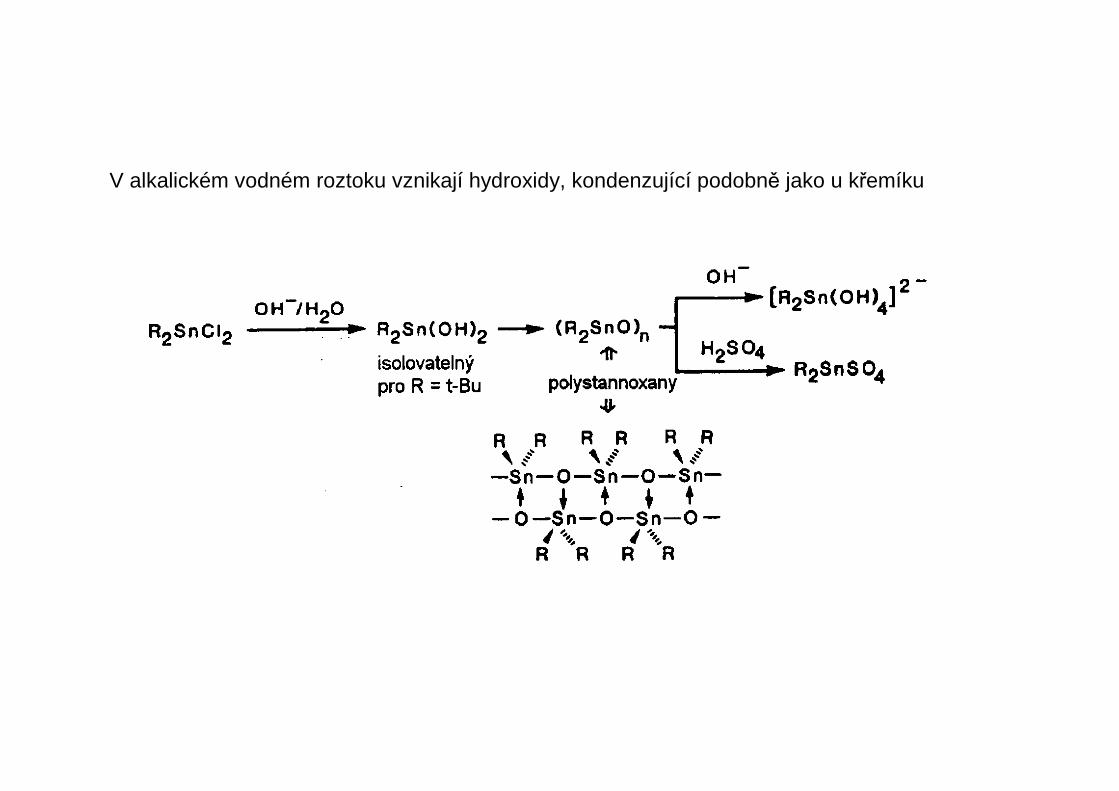
V alkalickém vodném roztoku vznikají hydroxidy, kondenzující podobně jako u křemíku
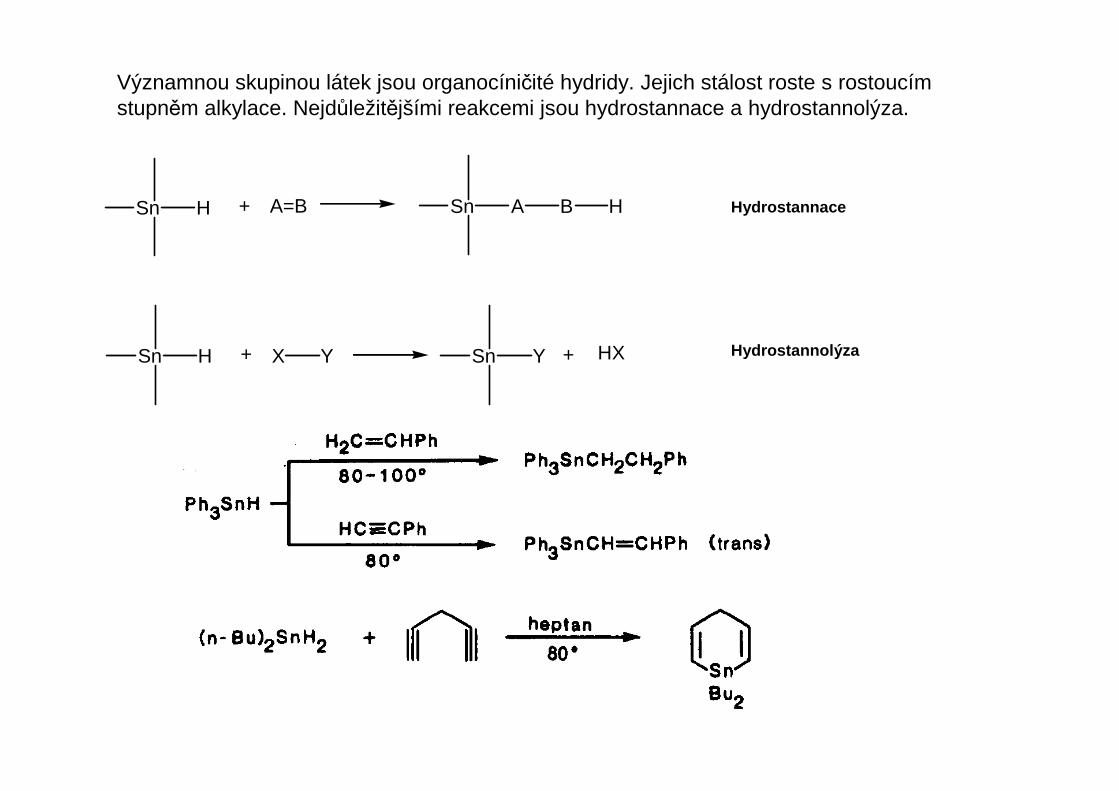
Významnou skupinou látek jsou organocíničité hydridy. Jejich stálost roste s rostoucím stupněm alkylace. Nejdůležitějšími reakcemi jsou hydrostannace a hydrostannolýza.
Hydrostannace
Hydrostannolýza
Sn H + A=B Sn A B H
Sn H + X Y Sn Y + HX
Při hydrostannolýze se v důsledku nízké polarity vazby Sn-H vodík přenáší podle vlastnostísubstrátu jako radikál, kation nebo anion.
R3SnH
4 R3SnH
2 R3SnH
+ MeCOOH R3SnOOCMe
+ Ti(NR'2)4 (R3Sn)4Ti + 4 HNR'2
+ R'2Hg (R3Sn)2Hg + 2 R'H
H
H
H
Přenos
Hydrostannolýza je univerzální metodou konverze organických halogenidů na uhlovodíky. Je to radikálová reakce.
R3SnH R3Sn + H
R3Sn + R'X R3SnX + R'
R' + R3SnH R'H + R3Sn
Toxicita organocíničitých sloučenin vzrůstá s rostoucím stupněm alkylace a klesá s rostoucídélkou alkylového řetězce. Nejjedovatější jsou látky, které mohou přecházet na kation R3Sn+, naproti tomu Bu2Sn(SCH2COO-iso-C8H17)2 je prakticky netoxický.Do netoxické skupiny patří i změkčovadla a stabilizátory plastů typu R2SnCl2.Sloučeniny typu R3SnX slouží jako fungicidy a pesticidy v zemědělství a ochraně staveb. Biologicky se degradují na netoxický hydratovaný oxid cíničitý.
Organické sloučeniny olova
Příprava
PbCl2 + 2 MeMgI Me4Pb + Pb
Pb(OAc)4 + 4 RMgCl R4Pb + 4 MgClOAc
2 MeI + Pb Me2PbI2
R3PbCl + LiR' R3PbR' + LiCl
R3PbH + H2C=CHR' R3PbCH2CH2R'
6 RMgX + 3 PbCl2 R3PbPbR3 + Pb + 6 MgXCl- 20 °C
V průmyslkovém měřítku se tetraethylolovo vyrábí reakcí ethylchloridu se slitinou sodíku a olova při 110 °C za katalýzy slou čeninami Cd.Jedovaté tetraethylolovo se používalo desítky let jako antidetonační přísada do benzínů, protože je jednak v uhlovodících rozpustné a za druhé se rozkládá na radikály blokujícísamovolné radikálové reakce. Dnes je nahrazeno methyl-terc-butyletherem.
Skupina 15 (5. hlavní podskupina): P, ostatní prvky
Skupina 16 (6. hlavní podskupina): Se, Te
Skupina 11 (1. vedlejší podskupina): Cu, Ag, Au
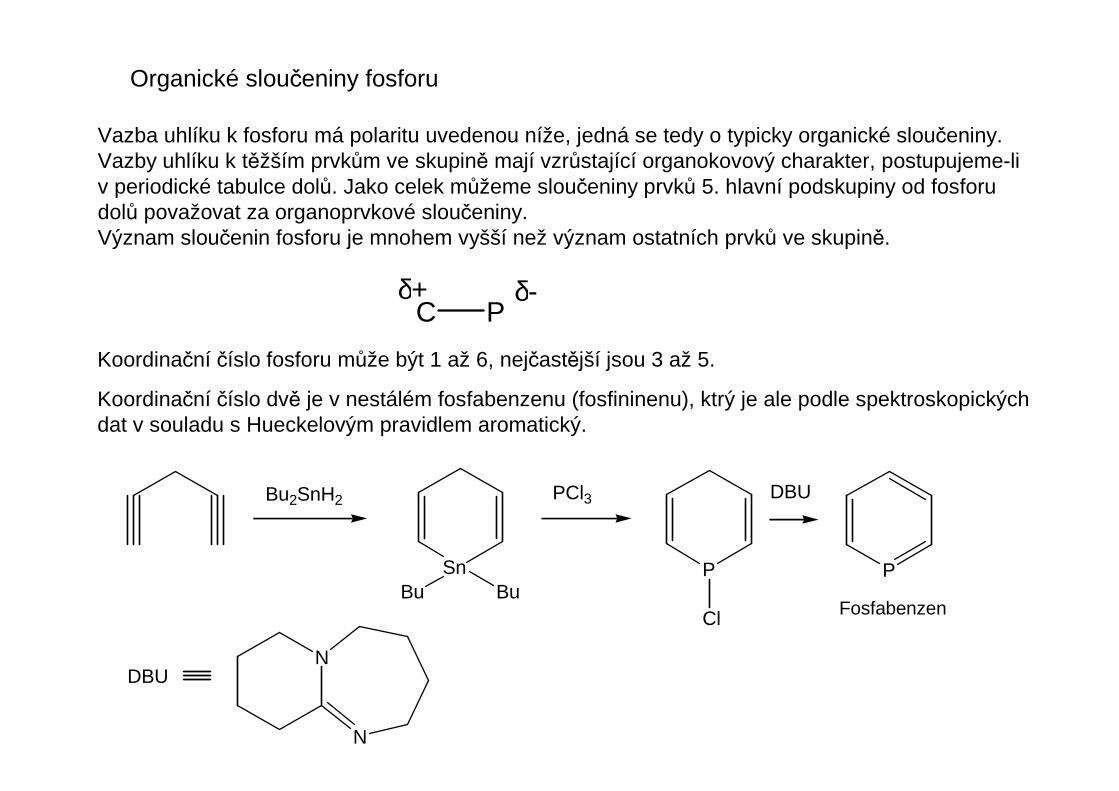
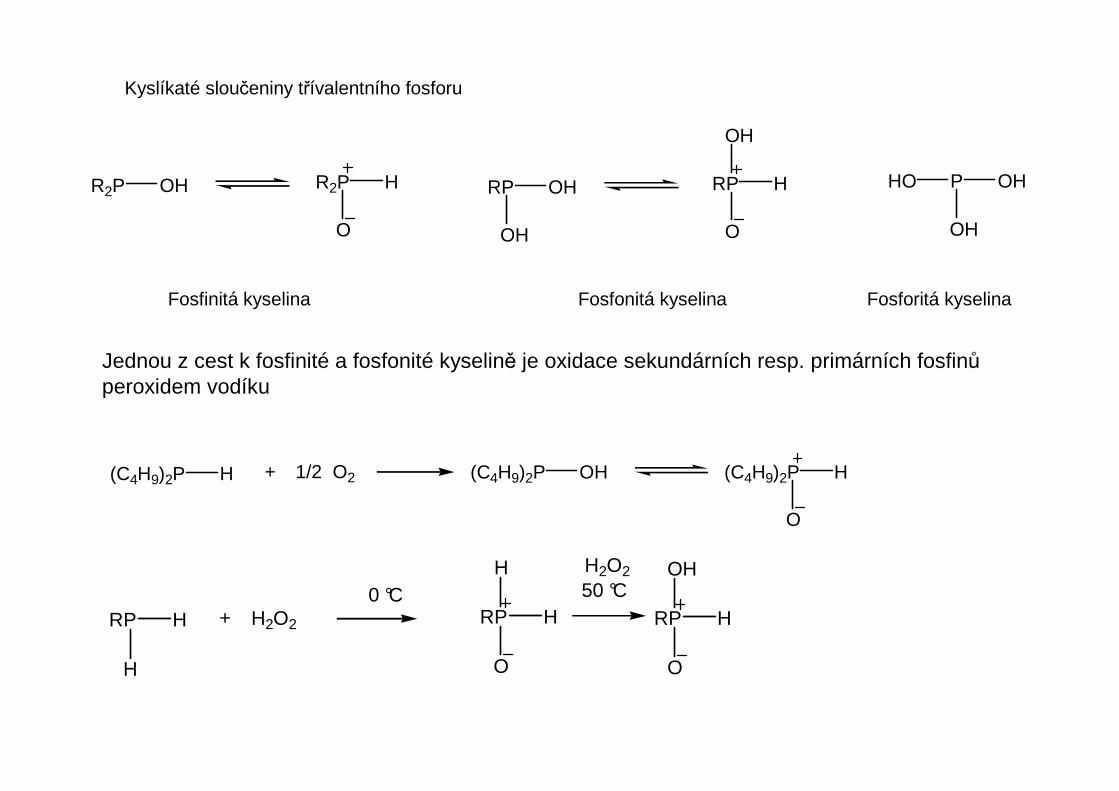
Organické sloučeniny fosforu
Vazba uhlíku k fosforu má polaritu uvedenou níže, jedná se tedy o typicky organické sloučeniny.Vazby uhlíku k těžším prvkům ve skupině mají vzrůstající organokovový charakter, postupujeme-liv periodické tabulce dolů. Jako celek můžeme sloučeniny prvků 5. hlavní podskupiny od fosforudolů považovat za organoprvkové sloučeniny.Význam sloučenin fosforu je mnohem vyšší než význam ostatních prvků ve skupině.
C Pδ-δ+
Koordinační číslo fosforu může být 1 až 6, nejčastější jsou 3 až 5.
Koordinační číslo dvě je v nestálém fosfabenzenu (fosfininenu), ktrý je ale podle spektroskopickýchdat v souladu s Hueckelovým pravidlem aromatický.
Bu2SnH2
SnBu Bu
PCl3
P
Cl
DBU
P
N
N
DBU
Fosfabenzen
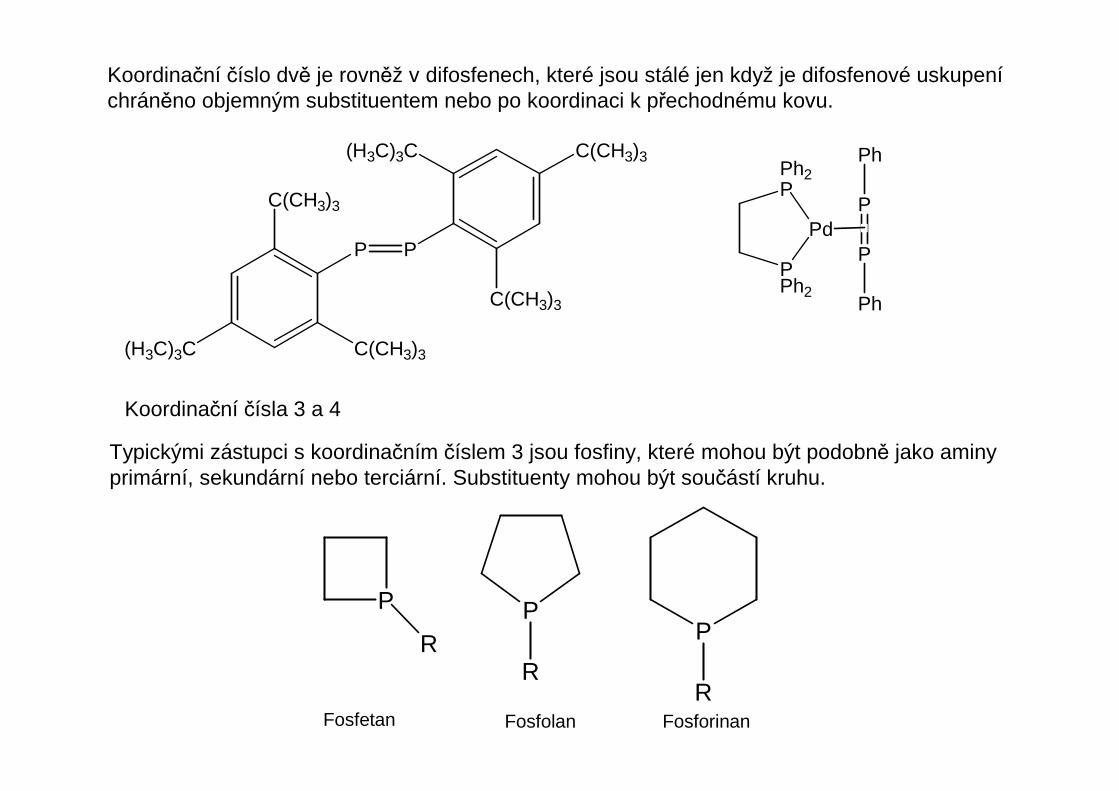
Koordinační číslo dvě je rovněž v difosfenech, které jsou stálé jen když je difosfenové uskupeníchráněno objemným substituentem nebo po koordinaci k přechodnému kovu.
P P
(H3C)3C
C(CH3)3
C(CH3)3
C(CH3)3
C(CH3)3(H3C)3C
PPh2
Pd
Ph2P
P
P
Ph
Ph
Koordinační čísla 3 a 4
Typickými zástupci s koordinačním číslem 3 jsou fosfiny, které mohou být podobně jako aminyprimární, sekundární nebo terciární. Substituenty mohou být součástí kruhu.
P PP
RR
R
Fosfetan Fosfolan Fosforinan
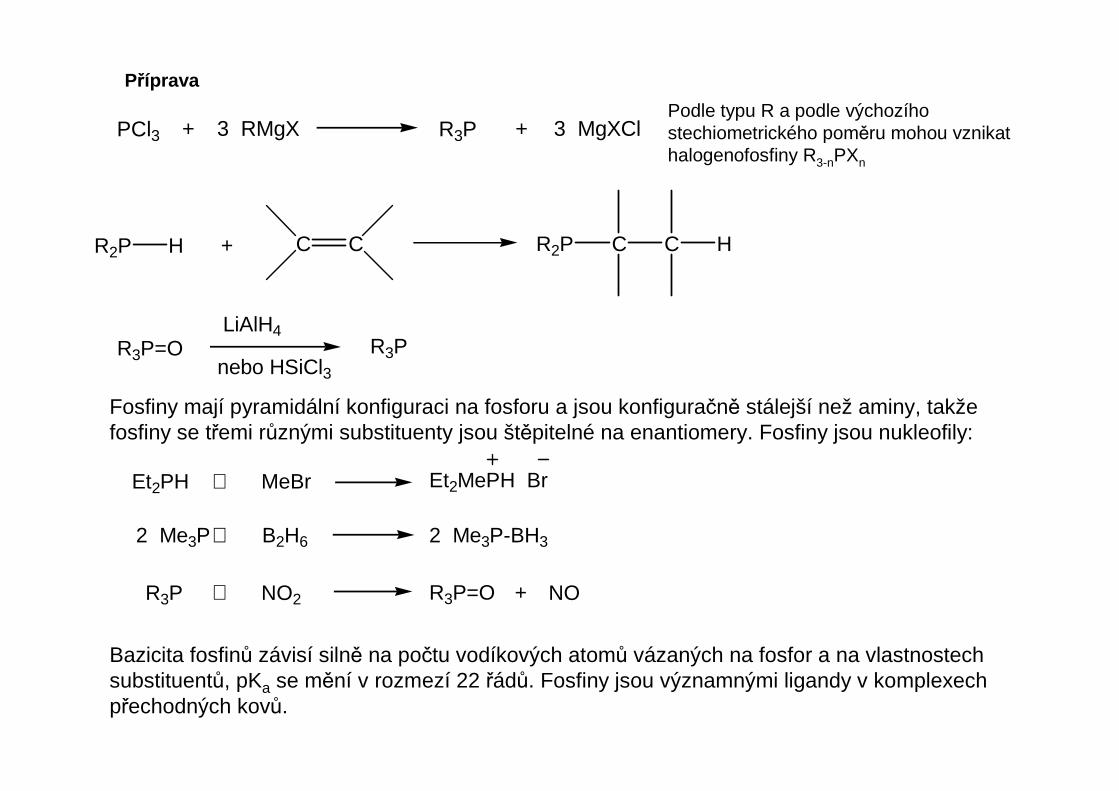
Příprava
PCl3 + 3 RMgX R3P + 3 MgXClPodle typu R a podle výchozíhostechiometrického poměru mohou vznikathalogenofosfiny R3-nPXn
C CR2P H + R2P C C H
R3P=OLiAlH4
nebo HSiCl3R3P
Fosfiny mají pyramidální konfiguraci na fosforu a jsou konfiguračně stálejší než aminy, takžefosfiny se třemi různými substituenty jsou štěpitelné na enantiomery. Fosfiny jsou nukleofily:
Et2PH + MeBr Et2MePH Br
2 Me3P+ B2H6 2 Me3P-BH3
R3P + NO2 R3P=O + NO
Bazicita fosfinů závisí silně na počtu vodíkových atomů vázaných na fosfor a na vlastnostechsubstituentů, pKa se mění v rozmezí 22 řádů. Fosfiny jsou významnými ligandy v komplexechpřechodných kovů.
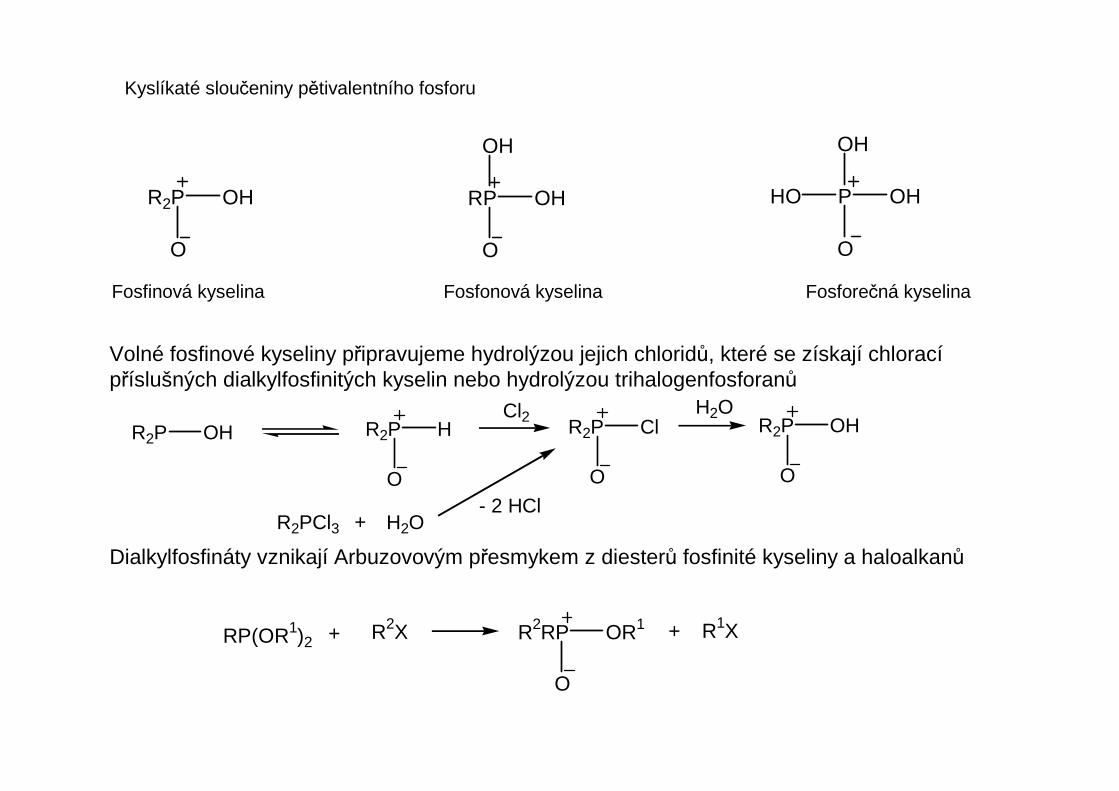
Fosfoniové soli obsahují tetraedricky koordinovaný (koordinační číslo 4) atom fosforu. Jsou narozdíl od amoniových solí konfiguračně stálé podobně jako fosfiny. V alkalickém prostředí se štěpívazba P-C, přičemž zpravidla odstupuje stericky nejnáročnější substituent.
R3PR' R3P=O + R'HHO
Fosfinoxidy R3-nHnP=O mohou být podobně jako fosfiny primární, sekundární a terciární. Mimojiné jsou prvním produktem oxidace fosfinů. Terciární fosfinoxidy jsou vůči další oxidaci stálé.V primárních a sekundárních fosfinoxidech jsou atomy vodíku vázané na fosfor slabě kyseléa reaktivní.
Jednou z cest k fosfinité a fosfonité kyselině je oxidace sekundárních resp. primárních fosfinůperoxidem vodíku
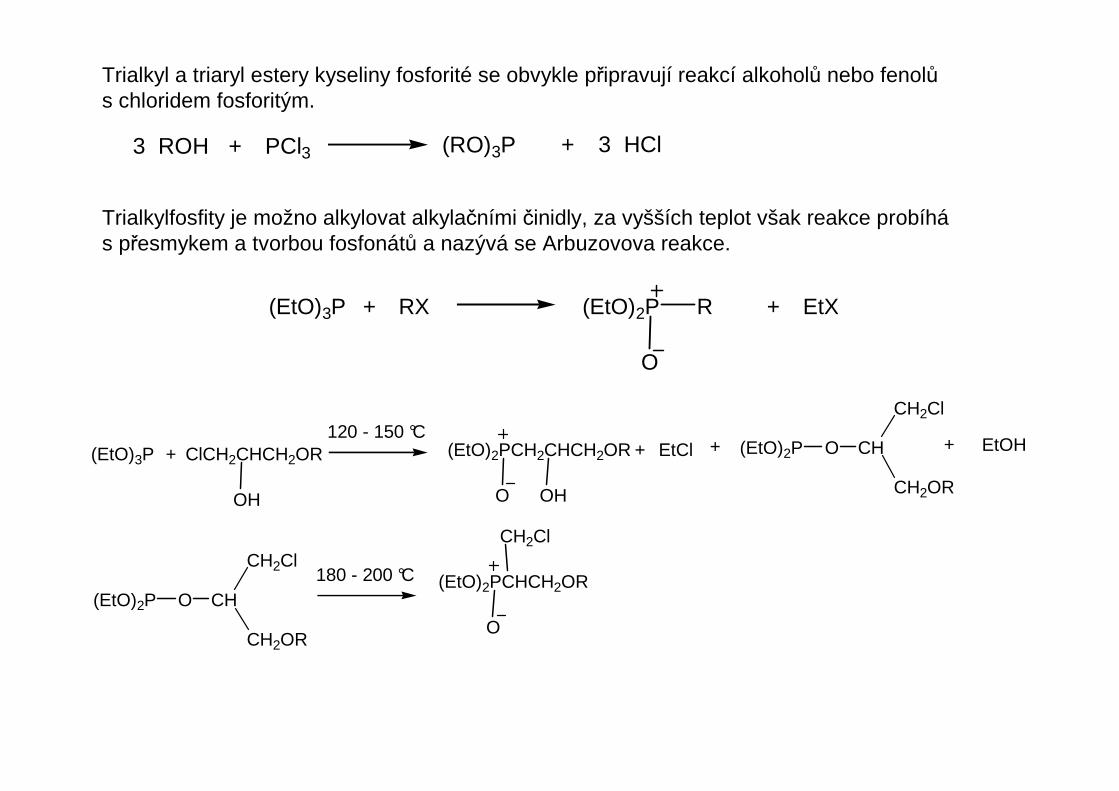
Trialkyl a triaryl estery kyseliny fosforité se obvykle připravují reakcí alkoholů nebo fenolůs chloridem fosforitým.
3 ROH + PCl3 (RO)3P + 3 HCl
Trialkylfosfity je možno alkylovat alkylačními činidly, za vyšších teplot však reakce probíhás přesmykem a tvorbou fosfonátů a nazývá se Arbuzovova reakce.
Volné fosfinové kyseliny připravujeme hydrolýzou jejich chloridů, které se získají chloracípříslušných dialkylfosfinitých kyselin nebo hydrolýzou trihalogenfosforanů
R2P OH R2P H
O
Cl2R2P Cl
O
H2OR2P OH
O
R2PCl3 + H2O- 2 HCl
Dialkylfosfináty vznikají Arbuzovovým přesmykem z diesterů fosfinité kyseliny a haloalkanů
RP(OR1)2 + R2X R2RP OR1
O
+ R1X
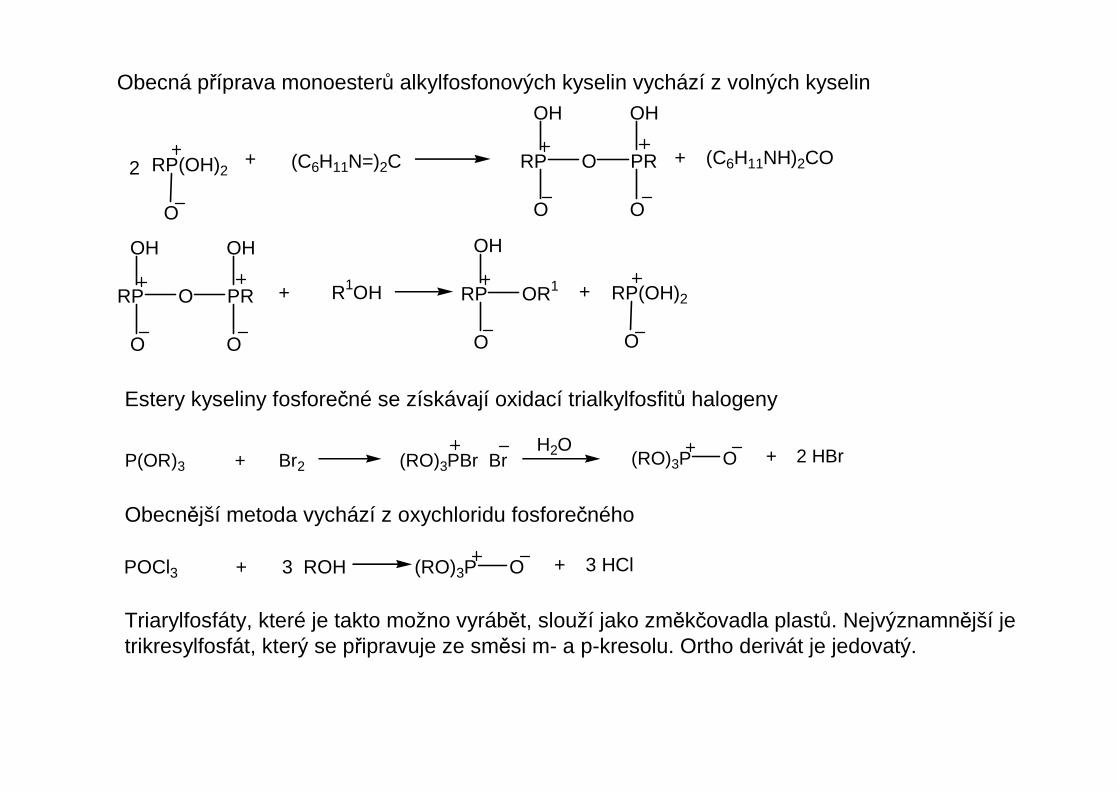
Obecná příprava monoesterů alkylfosfonových kyselin vychází z volných kyselin
RP(OH)2
O
2 + (C6H11N=)2C RP O
O
OH
PR
OH
O
+ (C6H11NH)2CO
RP O
O
OH
PR
OH
O
+ R1OH RP OR1
O
OH
+ RP(OH)2
O
Estery kyseliny fosforečné se získávají oxidací trialkylfosfitů halogeny
P(OR)3 + Br2 (RO)3PBr BrH2O (RO)3P O + 2 HBr
Obecnější metoda vychází z oxychloridu fosforečného
POCl3 + 3 ROH (RO)3P O + 3 HCl
Triarylfosfáty, které je takto možno vyrábět, slouží jako změkčovadla plastů. Nejvýznamnější jetrikresylfosfát, který se připravuje ze směsi m- a p-kresolu. Ortho derivát je jedovatý.
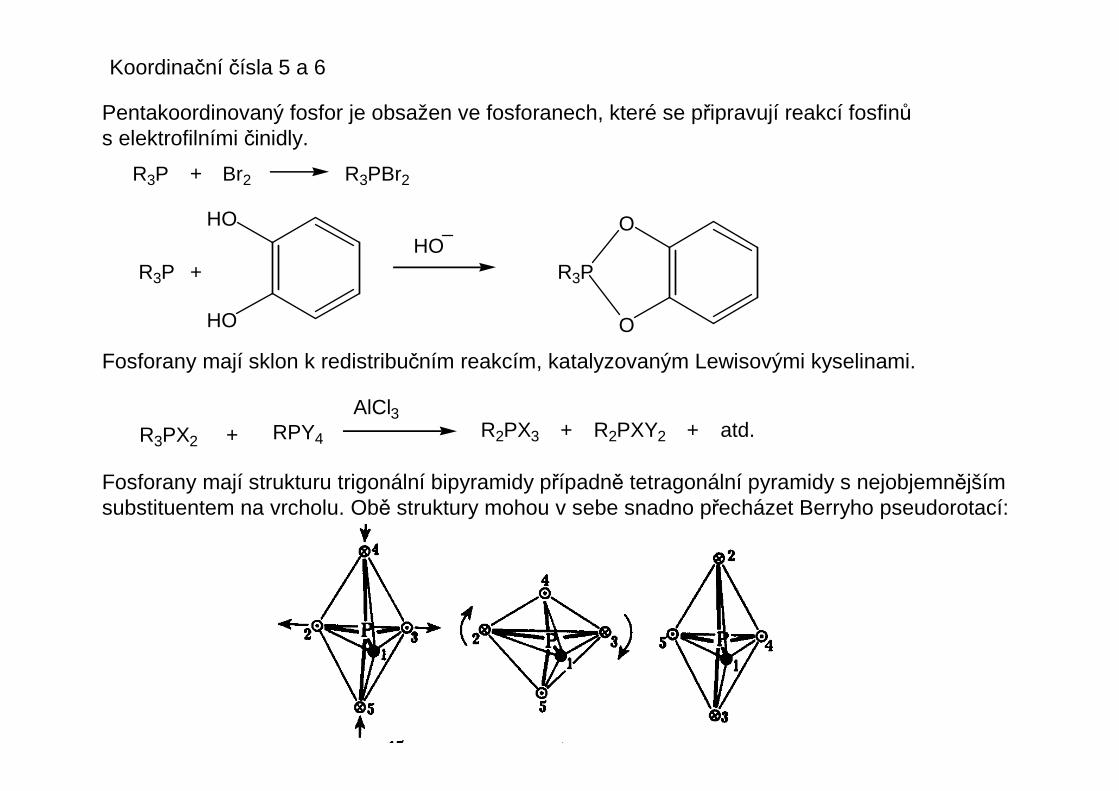
Koordinační čísla 5 a 6
Pentakoordinovaný fosfor je obsažen ve fosforanech, které se připravují reakcí fosfinůs elektrofilními činidly.
R3P + Br2 R3PBr2
R3P +
HO
HO
HOO
O
R3P
Fosforany mají sklon k redistribučním reakcím, katalyzovaným Lewisovými kyselinami.
R3PX2 + RPY4
AlCl3R2PX3 + R2PXY2 + atd.
Fosforany mají strukturu trigonální bipyramidy případně tetragonální pyramidy s nejobjemnějšímsubstituentem na vrcholu. Obě struktury mohou v sebe snadno přecházet Berryho pseudorotací:
RCH
(C6H5)3P
C
R'
R''
O
P(C6H5)3RCH C O
R'
R''
+ RCH
(C6H5)3P
C
R'
R''
O
RCH C
R'
R'' + (C6H5)3P O
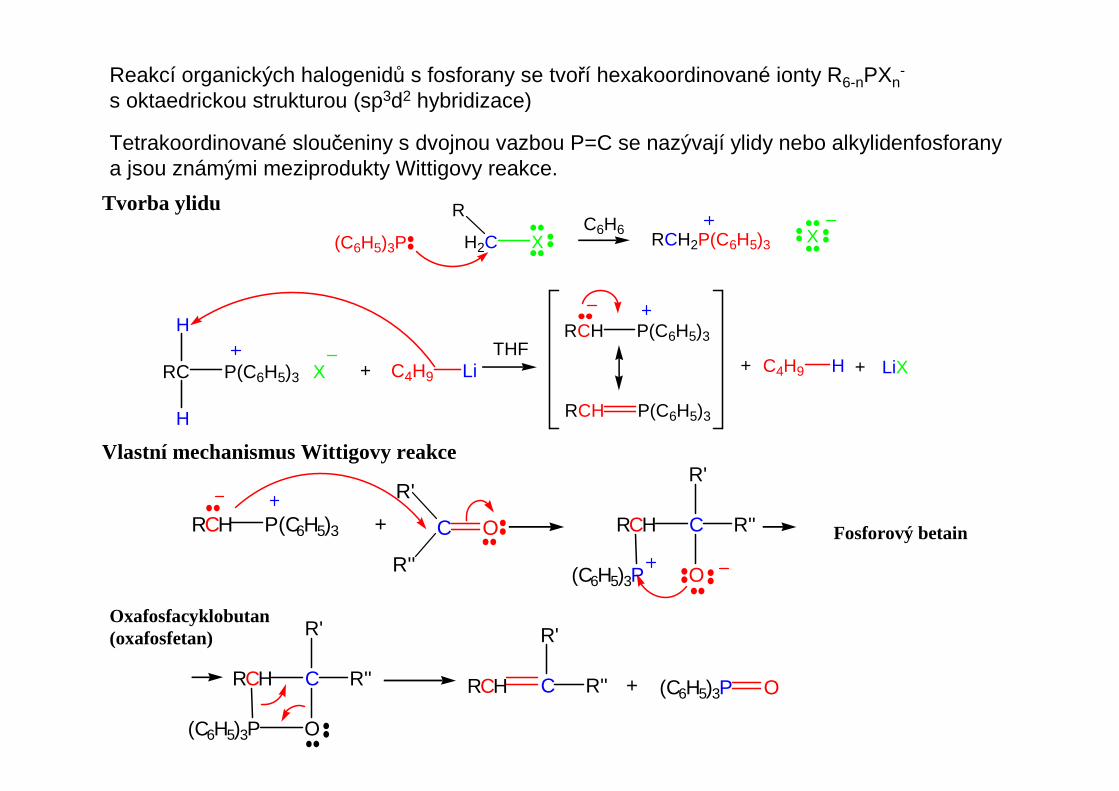
Reakcí organických halogenidů s fosforany se tvoří hexakoordinované ionty R6-nPXn-
s oktaedrickou strukturou (sp3d2 hybridizace)
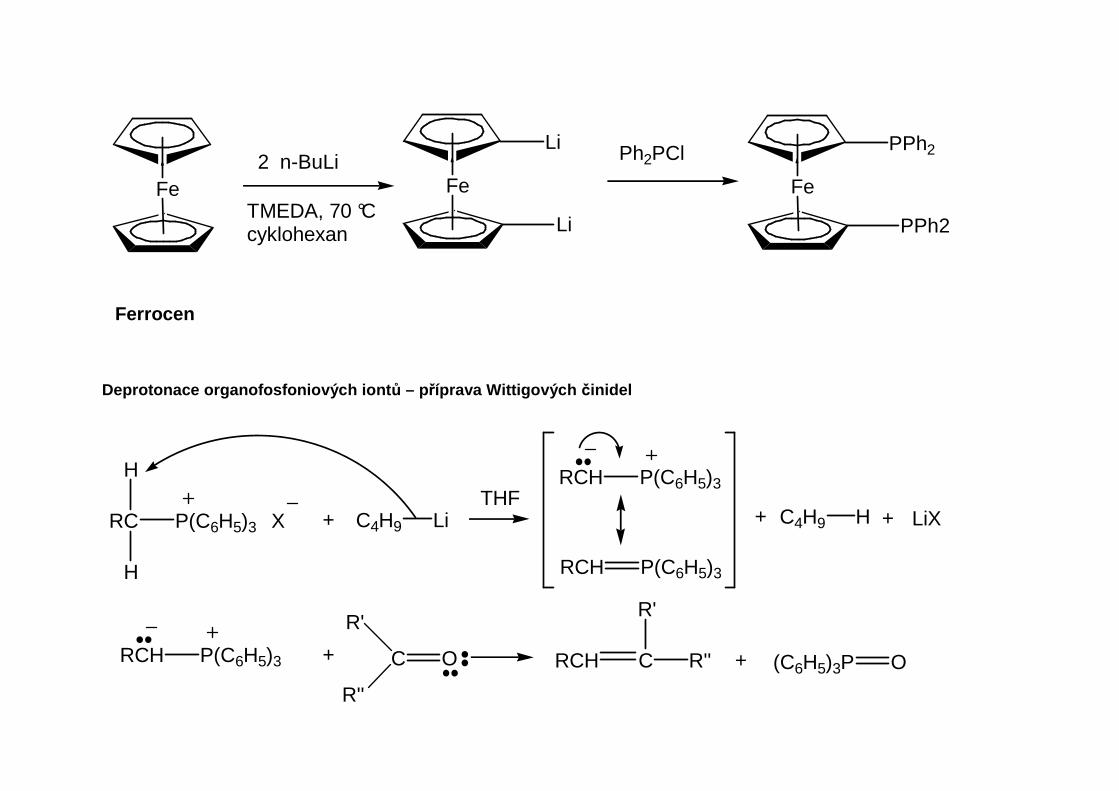
Tetrakoordinované sloučeniny s dvojnou vazbou P=C se nazývají ylidy nebo alkylidenfosforanya jsou známými meziprodukty Wittigovy reakce.
Tvorba ylidu
(C6H5)3P
R
H2C XC6H6
RCH2P(C6H5)3 X
P(C6H5)3 XRC
H
H
+ C4H9 LiTHF
P(C6H5)3RCH
P(C6H5)3RCH
+ C4H9 H + LiX
Oxafosfacyklobutan(oxafosfetan)
Vlastní mechanismus Wittigovy reakce
Fosforový betain
Organické sloučeniny arsenu, antimonu a bismutu
Organokovy těžších homologů 5. hlavní podskupiny jsou většinou analogické sloučeninámfosforu. Dobře známá je toxicita sloučenin arsenu. Použití sloučenin As v lékařství (Salvarsan)a v zemědělství (fungicidy) je dnes spíše historií. Objev možnosti biologické methylace anorganických látek (1897) kdysi poutal pozornost světové věřejnosti jako téměř detektivnípřípad: chronické otravy v nevětraných místnostech s tapetami barvenými svinibrodskou zelení (arseničnan měďnatý) byly způsobeny trimethylarsinem vznikajícím na vlhkém podkladu působením jistého druhu plísně.
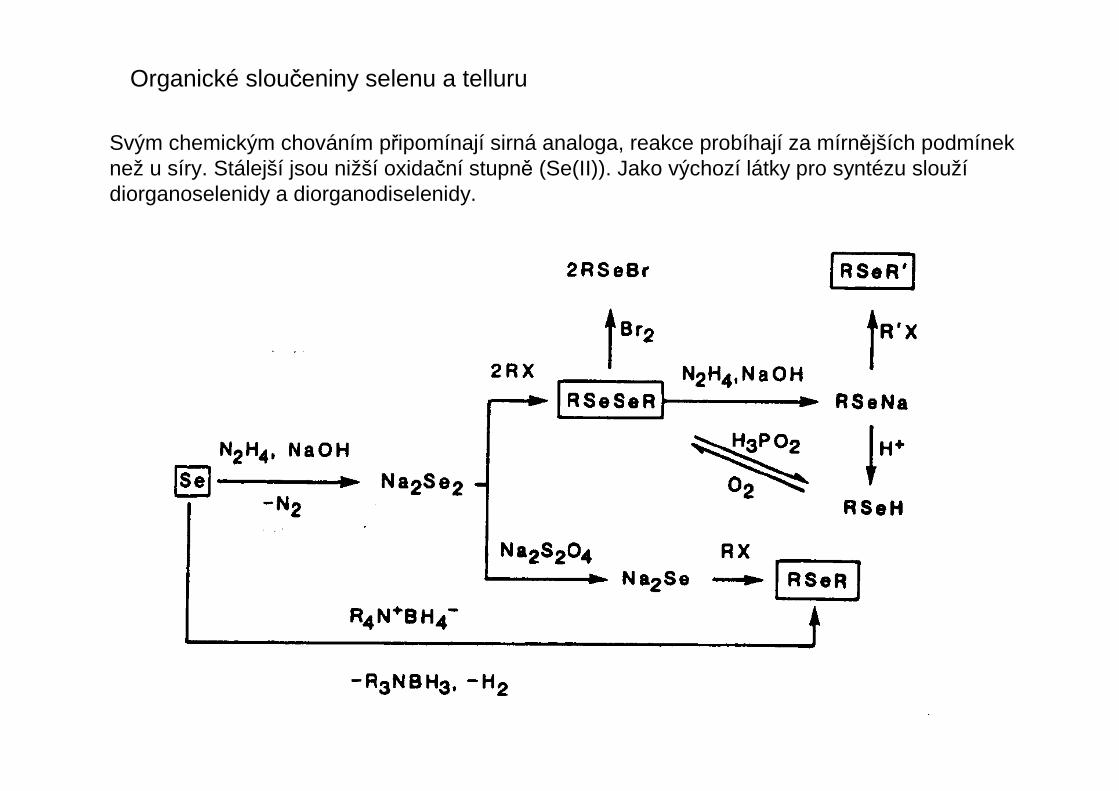
Organické sloučeniny selenu a telluru
Svým chemickým chováním připomínají sirná analoga, reakce probíhají za mírnějších podmíneknež u síry. Stálejší jsou nižší oxidační stupně (Se(II)). Jako výchozí látky pro syntézu sloužídiorganoselenidy a diorganodiselenidy.
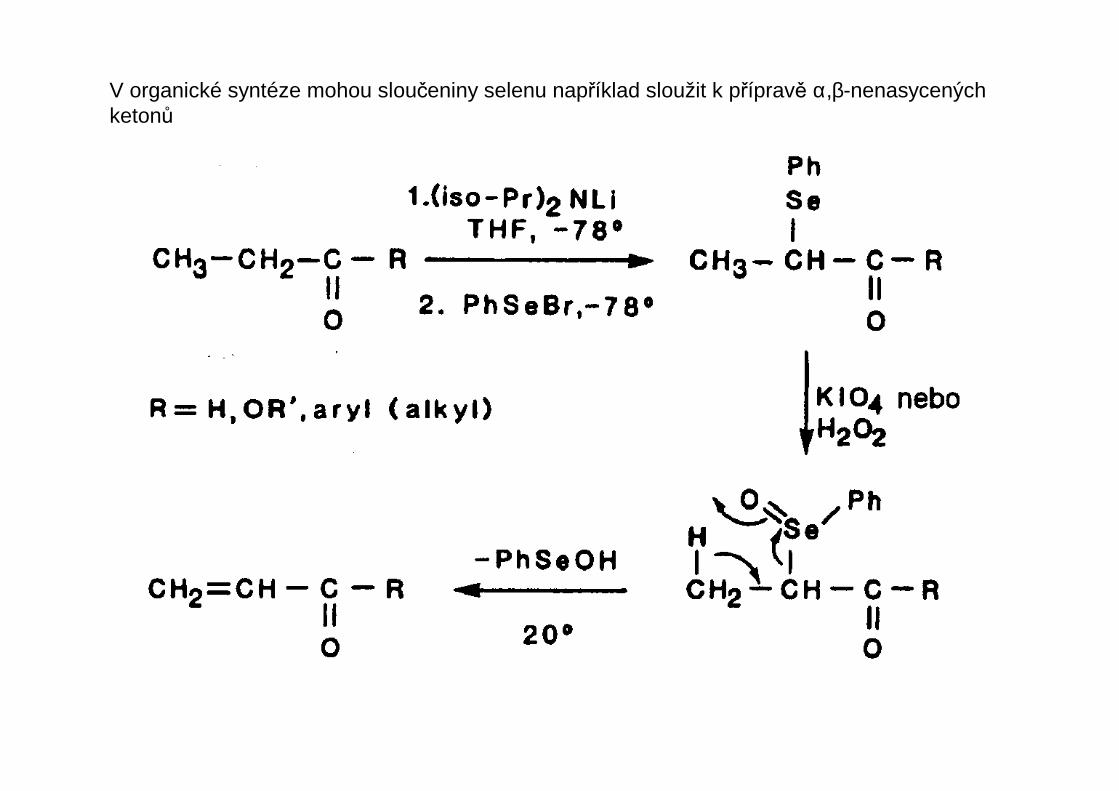
V organické syntéze mohou sloučeniny selenu například sloužit k přípravě α,β-nenasycenýchketonů
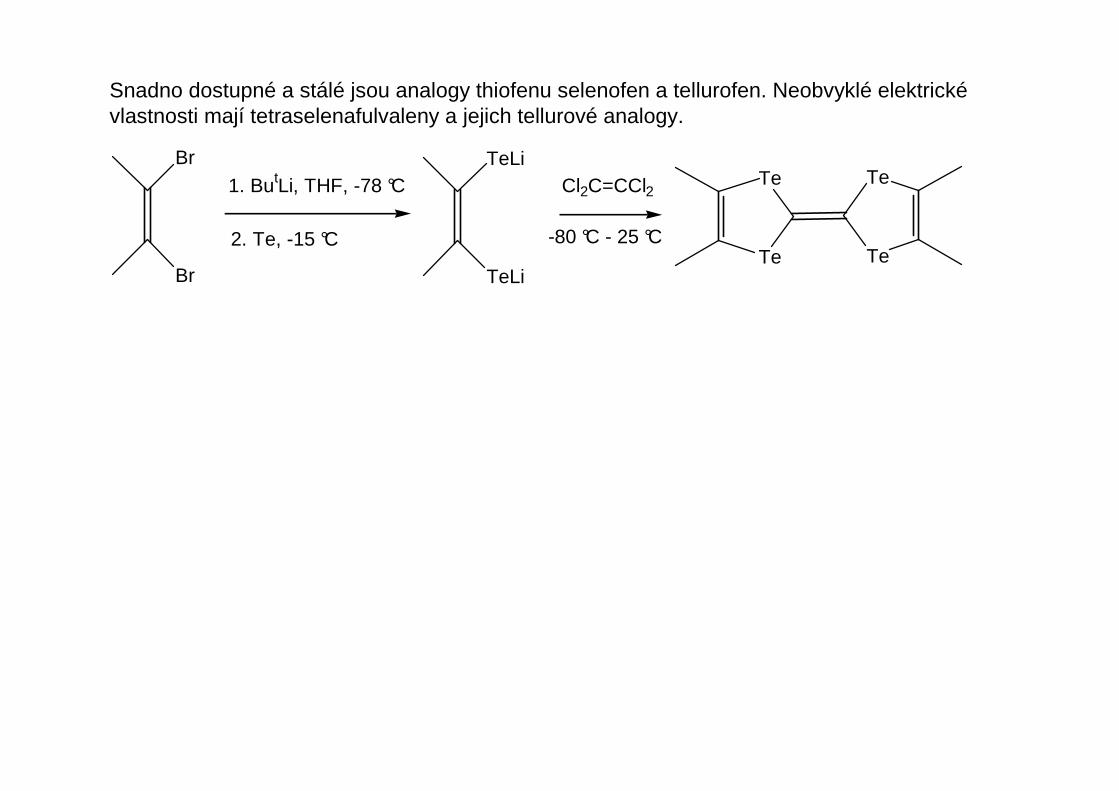
Snadno dostupné a stálé jsou analogy thiofenu selenofen a tellurofen. Neobvyklé elektrickévlastnosti mají tetraselenafulvaleny a jejich tellurové analogy.
Br
Br
1. ButLi, THF, -78 °C
2. Te, -15 °C
TeLi
TeLi
Cl2C=CCl2
-80 °C - 25 °CTe
Te Te
Te
Organické sloučeniny mědi a stříbra
Kovy měď, stříbro a zlato sice patří k přechodným prvkům, ale v mnoha sloučeninách majízaplněné d orbitaly, jejich chemie je proto podobná prvkům hlavních podskupin. Organokovováchemie Cu a Ag je omezena na jednomocenství, sloučeniny s π-vazbou budou diskutoványu příslušných ligandů.
Sloučeniny AgR jsou nestálejší než CuR a rozkládají se již za laboratorní teploty.
Příprava
Přes vžitý název diorganoměďnany nebodiorganokupráty nemají sloučeniny iontovoustrukturu: M+CuR2
-
AgNO3 + R4Pb AgR Ag + alkany + alkeny
2 LiAr + AgX LiAgAr2 + LiX
CuX + LiR CuR LiCuR2
LiREt2O (THF)
RC CH + [Cu(NH3)2]+
CuC CR + NH3 + NH4+
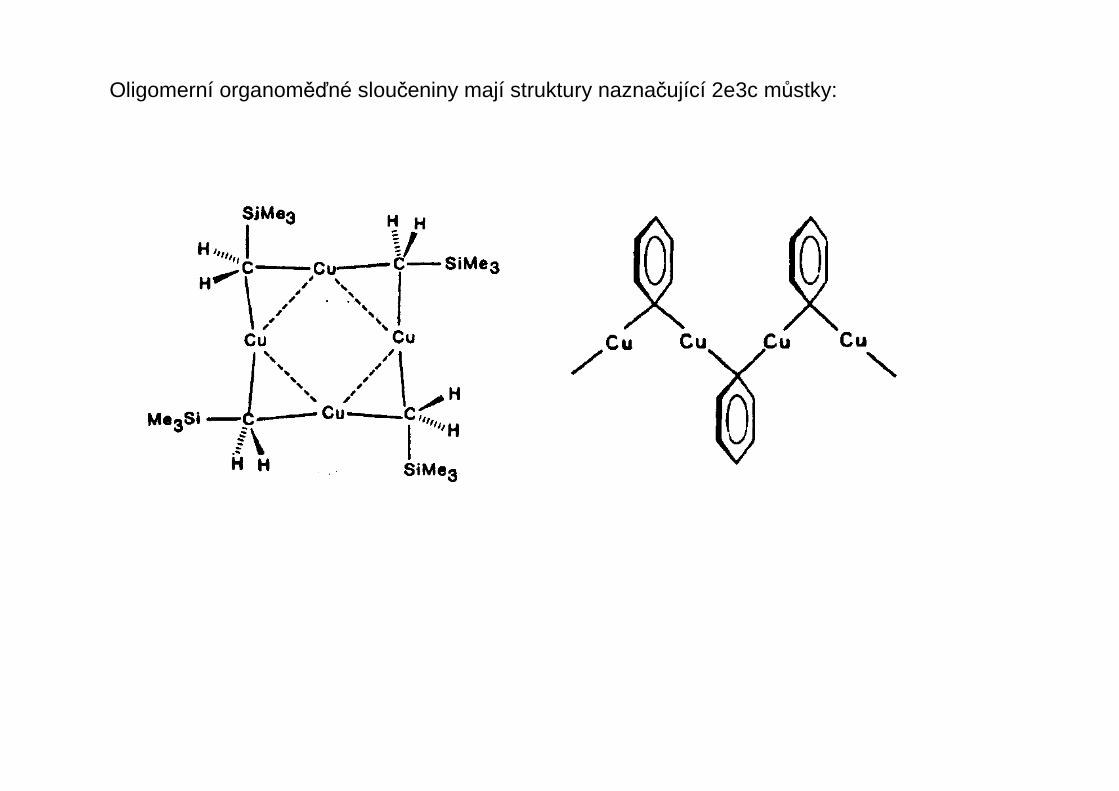
Oligomerní organoměďné sloučeniny mají struktury naznačující 2e3c můstky:
Reaktivita – použití v organické syntéze
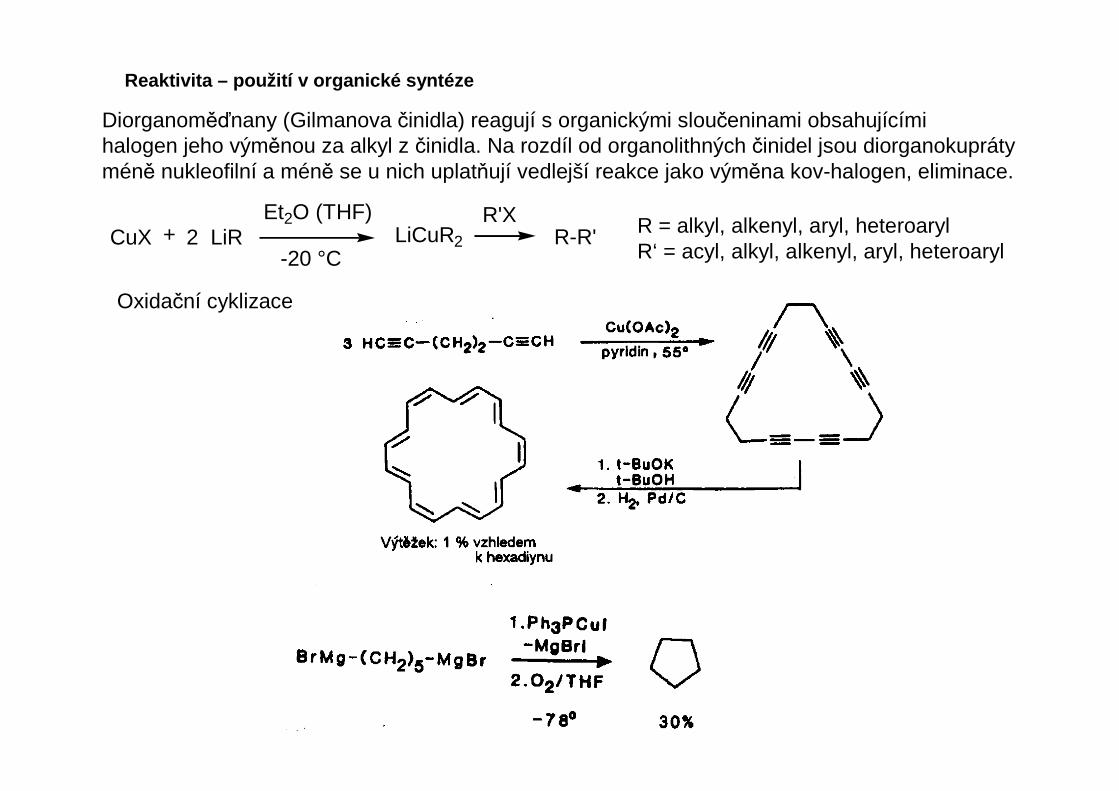
Diorganoměďnany (Gilmanova činidla) reagují s organickými sloučeninami obsahujícímihalogen jeho výměnou za alkyl z činidla. Na rozdíl od organolithných činidel jsou diorganokuprátyméně nukleofilní a méně se u nich uplatňují vedlejší reakce jako výměna kov-halogen, eliminace.
CuX + 2 LiR LiCuR2
R'XEt2O (THF)
-20 °CR-R' R = alkyl, alkenyl, aryl, heteroaryl
R‘ = acyl, alkyl, alkenyl, aryl, heteroaryl
Oxidační cyklizace
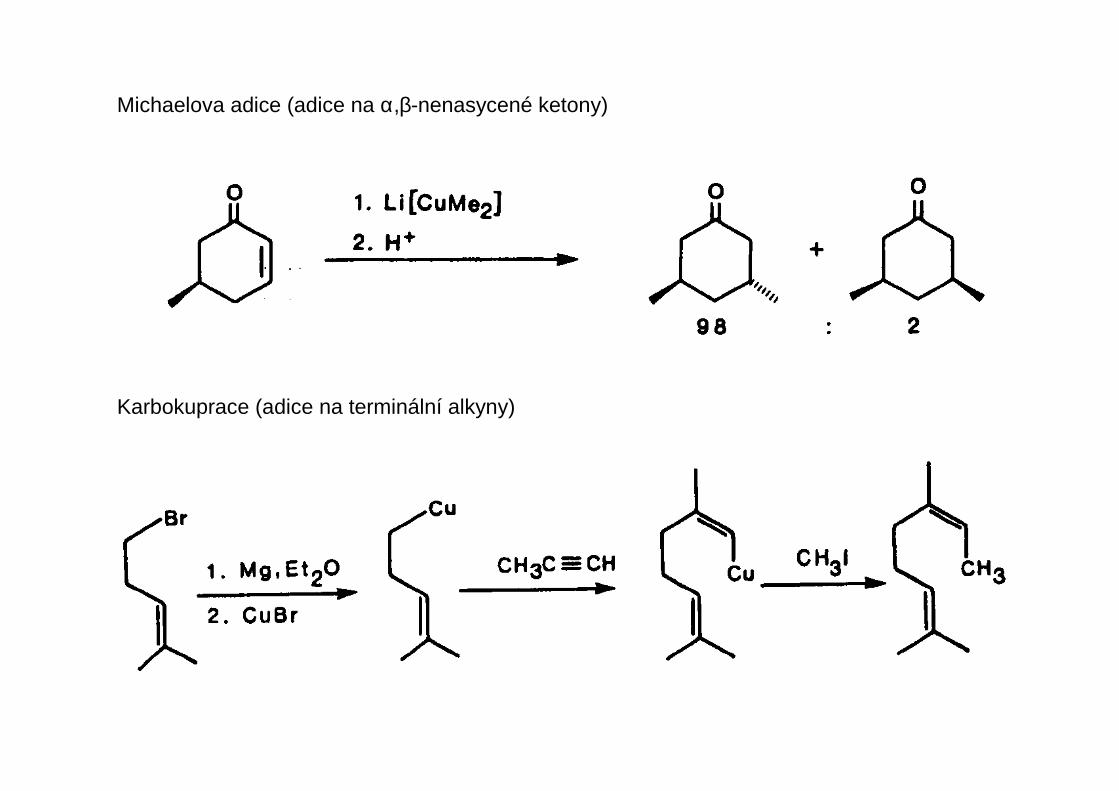
Michaelova adice (adice na α,β-nenasycené ketony)
Karbokuprace (adice na terminální alkyny)
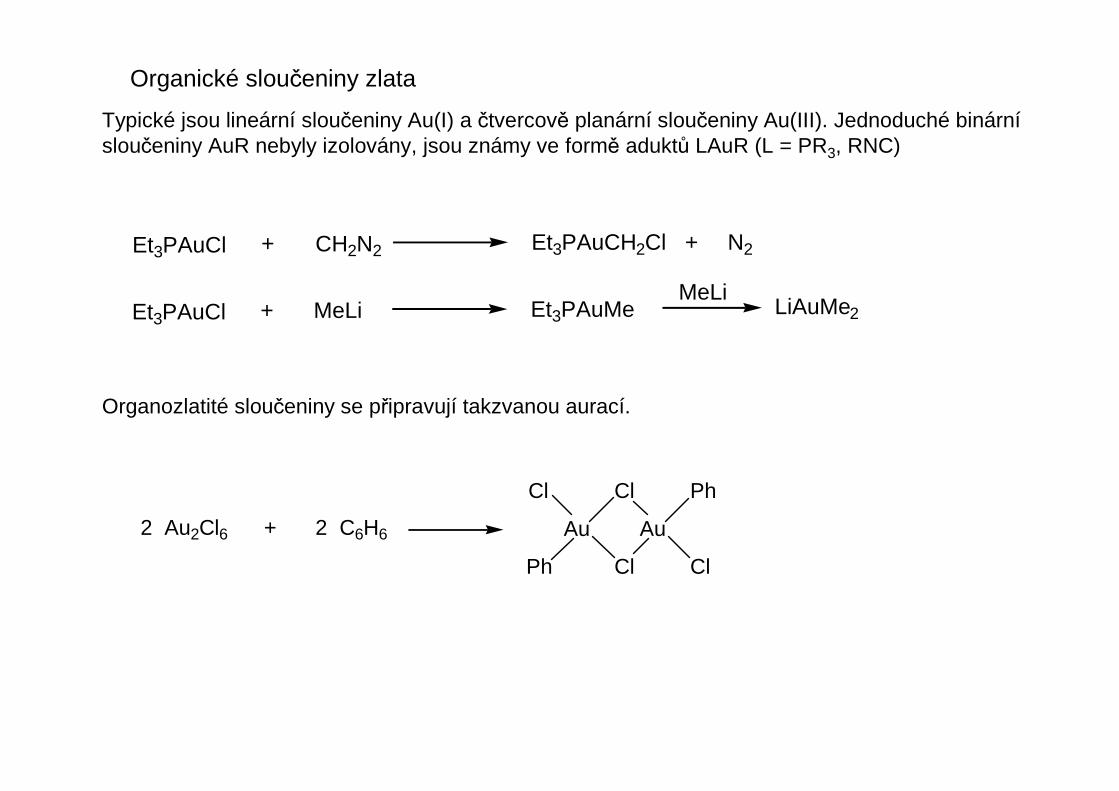
Organické sloučeniny zlata
Typické jsou lineární sloučeniny Au(I) a čtvercově planární sloučeniny Au(III). Jednoduché binárnísloučeniny AuR nebyly izolovány, jsou známy ve formě aduktů LAuR (L = PR3, RNC)
Organozlatité sloučeniny se připravují takzvanou aurací.
Et3PAuCl + CH2N2 Et3PAuCH2Cl + N2
Et3PAuCl + MeLi Et3PAuMe LiAuMe2MeLi
2 Au2Cl6 + 2 C6H6 Au
Cl
Au
Cl Ph
Cl
Cl
Ph
Vazebné možnosti d prvků, pravidlo 18 elektronů, výjimky
Sigma-donorové pi-akceptorové ligandyVinylyAlkynylyKarbeny a karbyny Karbonylové komplexy
Organokovové sloučeniny přechodných prvků
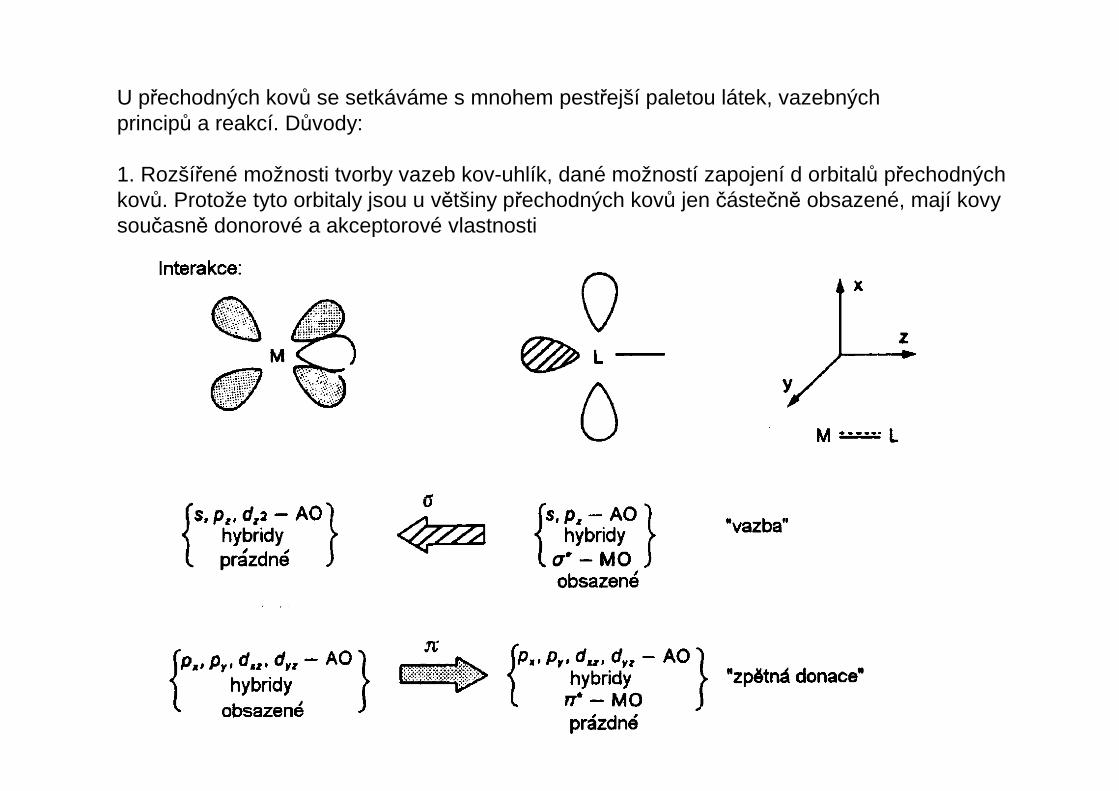
U přechodných kovů se setkáváme s mnohem pestřejší paletou látek, vazebnýchprincipů a reakcí. Důvody:
1. Rozšířené možnosti tvorby vazeb kov-uhlík, dané možností zapojení d orbitalů přechodnýchkovů. Protože tyto orbitaly jsou u většiny přechodných kovů jen částečně obsazené, mají kovysoučasně donorové a akceptorové vlastnosti
2. Schopnost snadné změny oxidačního čísla3. Schopnost změny koordinačního čísla a vytvoření volné koordinační pozice4. Velký výběr ligandů jak donorových tak akceptorových i těch, které mohou mít současnědonorové i akceptorové vlastnosti
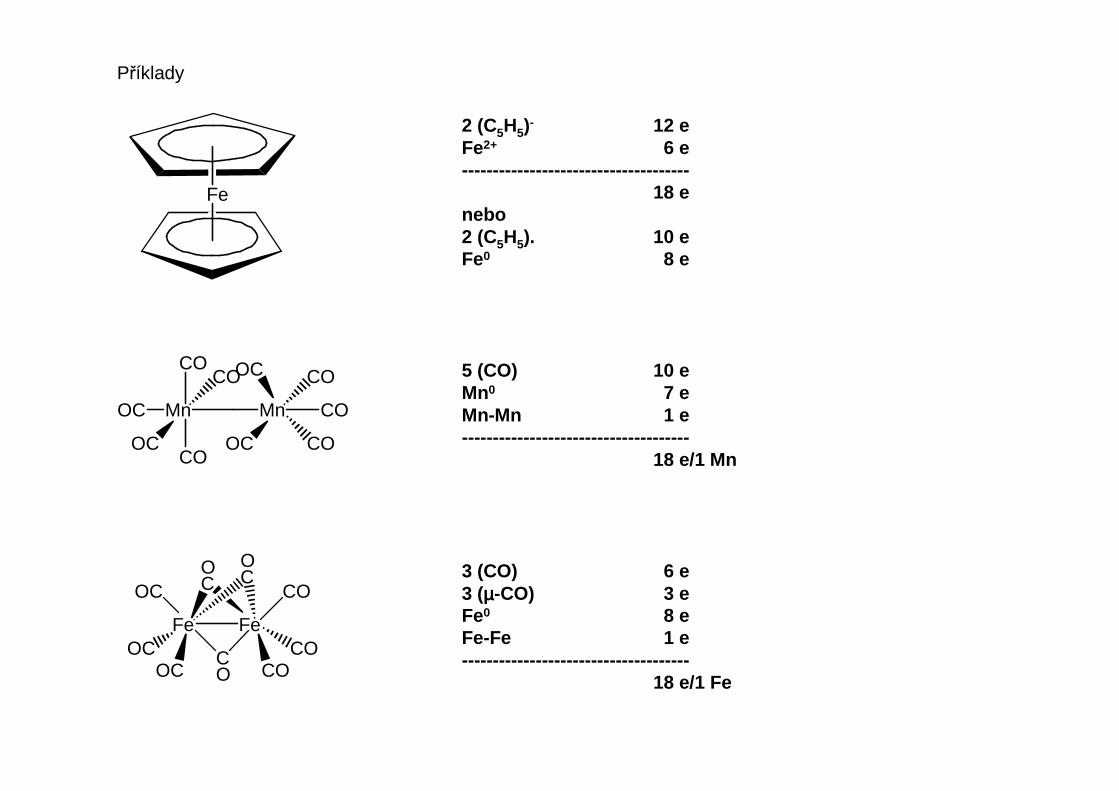
Pravidlo 1818 valenčních elektronů (Sidgwick, 1927, „efektivní atomové číslo“):Organokovové sloučeniny přechodných prvků jsou termodynamicky stálé tehdy,je-li součet d-elektronů kovu a elektronů poskytovaných ligandy roven 1818.
Pravidlo má přes velmi mnoho výjimek stále svoji platnost a užitečnost.
Platí, že:1. Nese-li posuzovaná částice náboj, za každý záporný náboj se jeden elektron přičte a za každý
kladný náboj jeden elektron odečte2. n-Násobná vazba kov-kov přispívá každému kovu n elektrony3. Můstkový ligand předává odpovídající podíl elektronů každému z kovů, které můstkuje.
Je-li podíl neceločíselný, sečtou se elektrony pro celou molekulu a součet se dělí počtemkovových atomů.
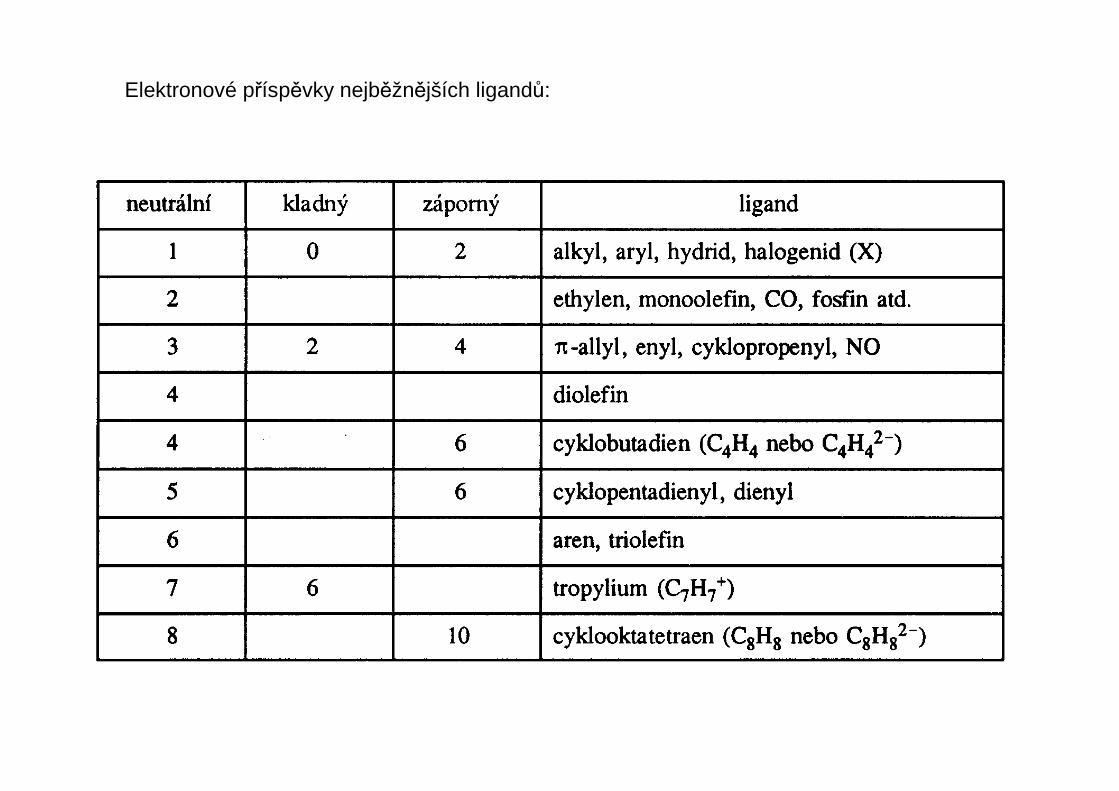
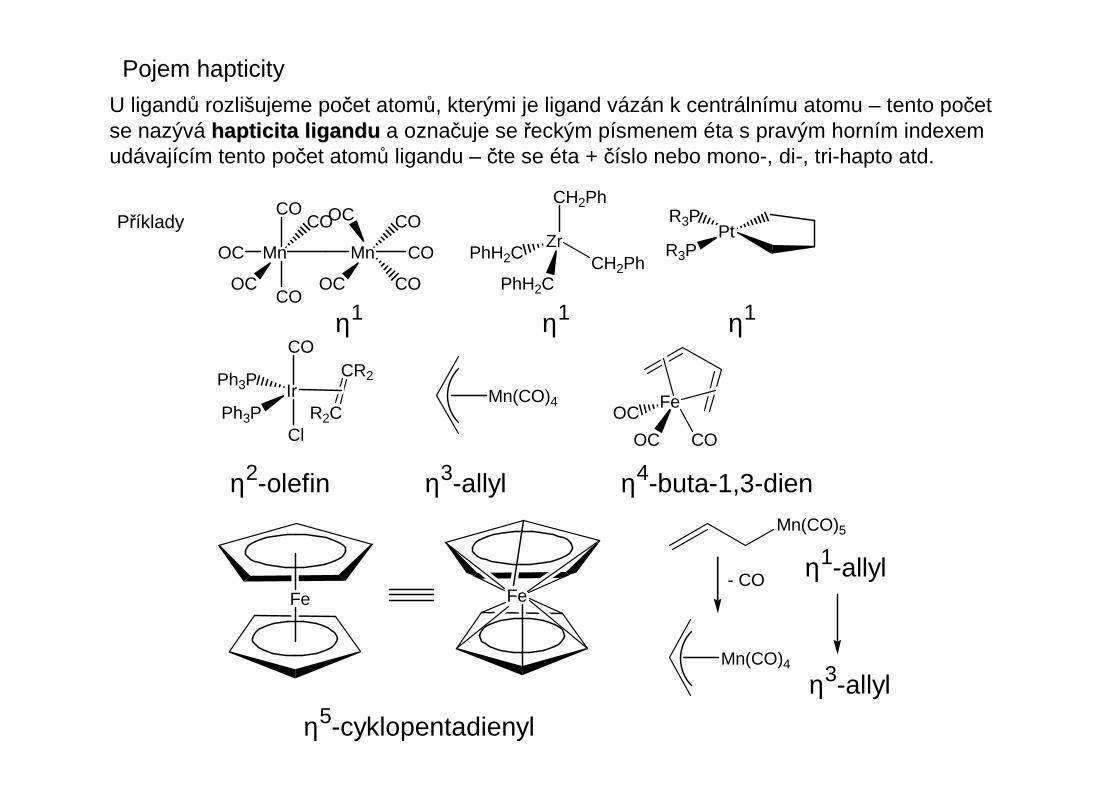
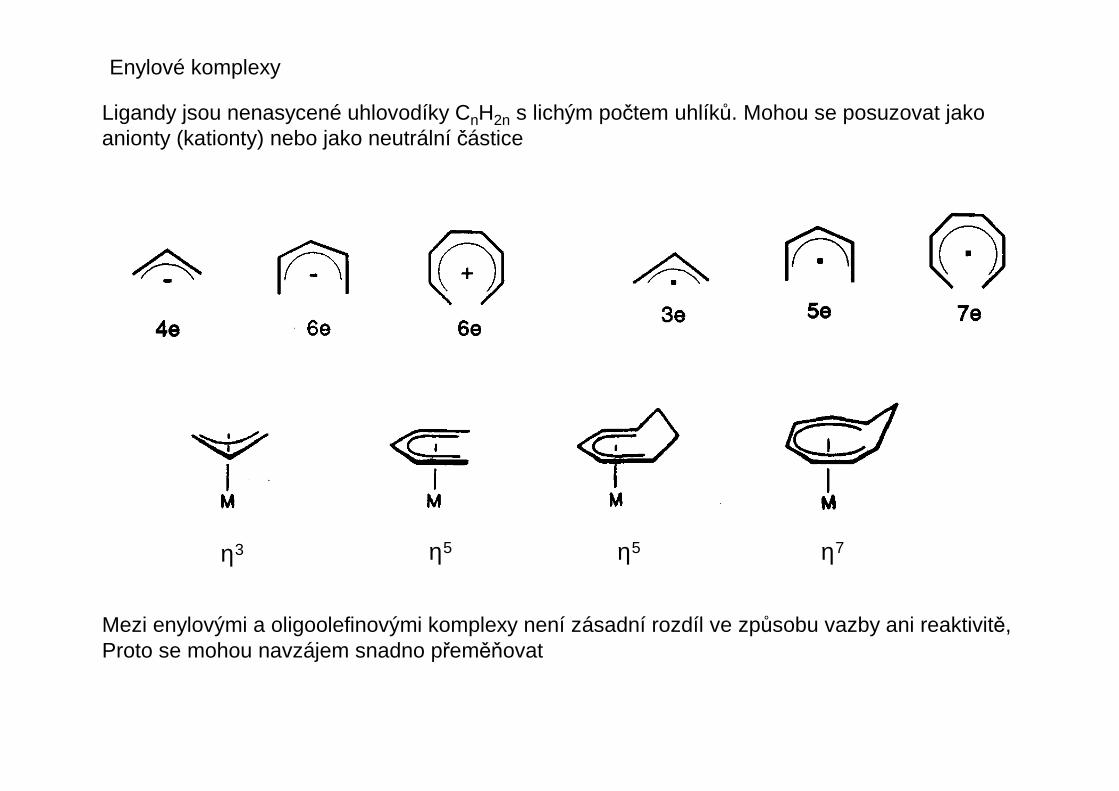
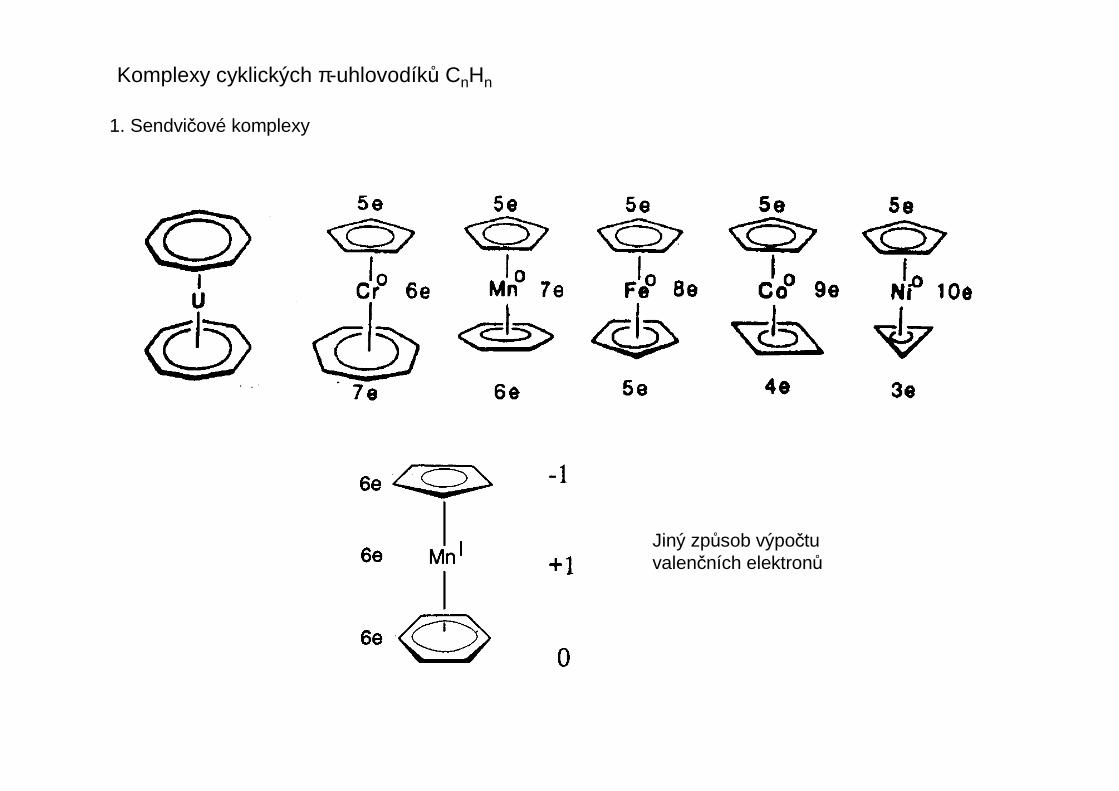
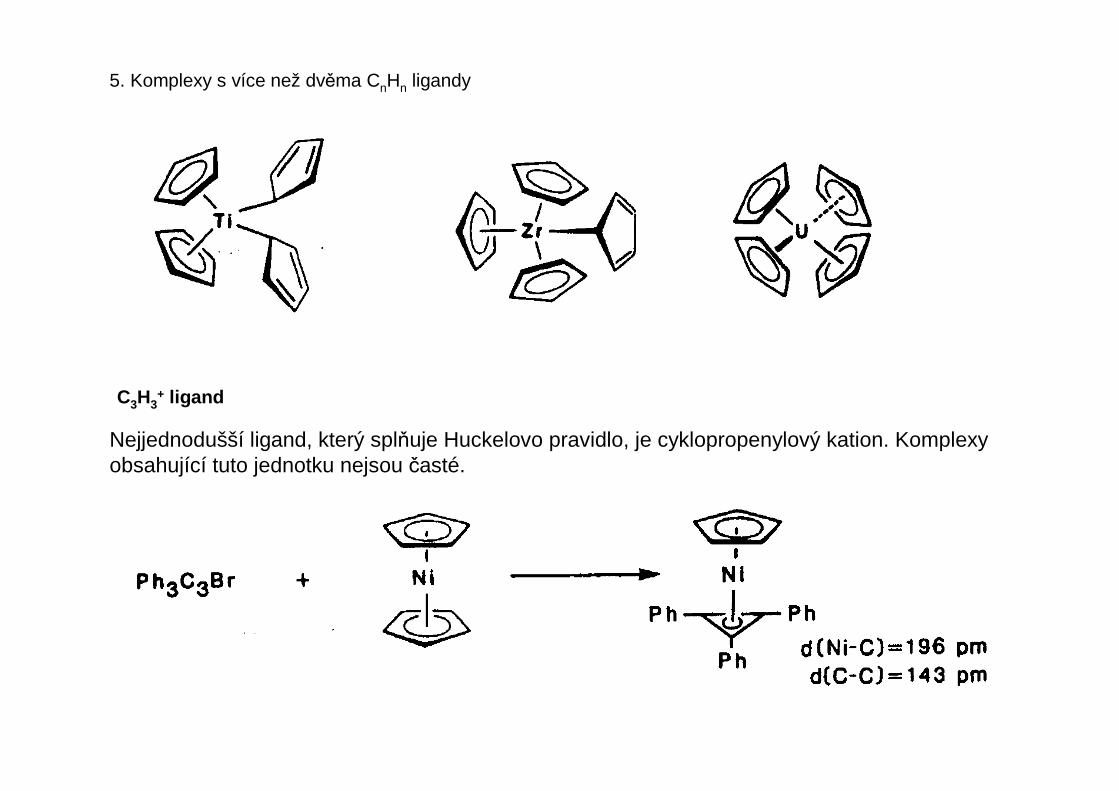
U ligandů rozlišujeme počet atomů, kterými je ligand vázán k centrálnímu atomu – tento početse nazývá hapticita liganduhapticita ligandu a označuje se řeckým písmenem éta s pravým horním indexemudávajícím tento počet atomů ligandu – čte se éta + číslo nebo mono-, di-, tri-hapto atd.
Příklady
Fe
OC Mn
CO
CO
Mn CO
CO
CO
OC
OCOC
COCH2Ph
ZrCH2Ph
PhH2C
PhH2CPt
R3P
R3P
Fe
Fe
COOC
OCIr
Ph3P
Ph3P
CO
Cl
CR2
R2CMn(CO)4
Mn(CO)5
- CO
Mn(CO)4
η1 η1 η1
η2-olefin η3-allyl η4-buta-1,3-dien
η5-cyklopentadienyl
η1-allyl
η3-allyl
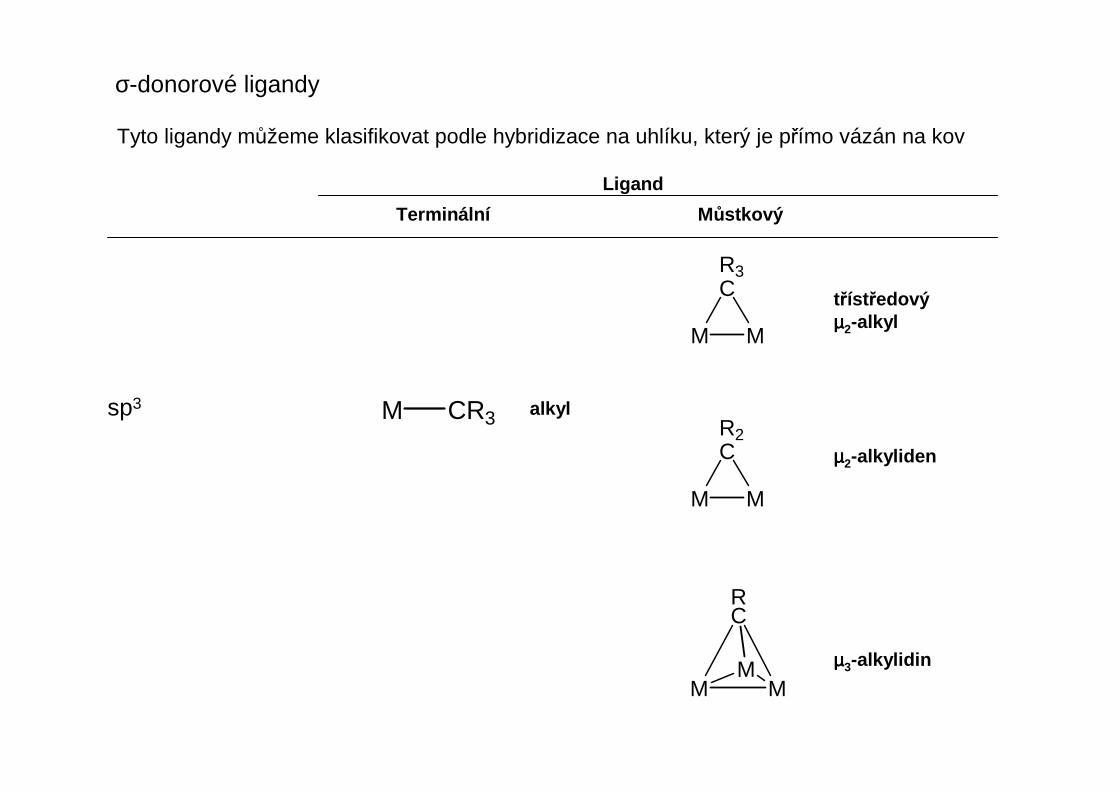
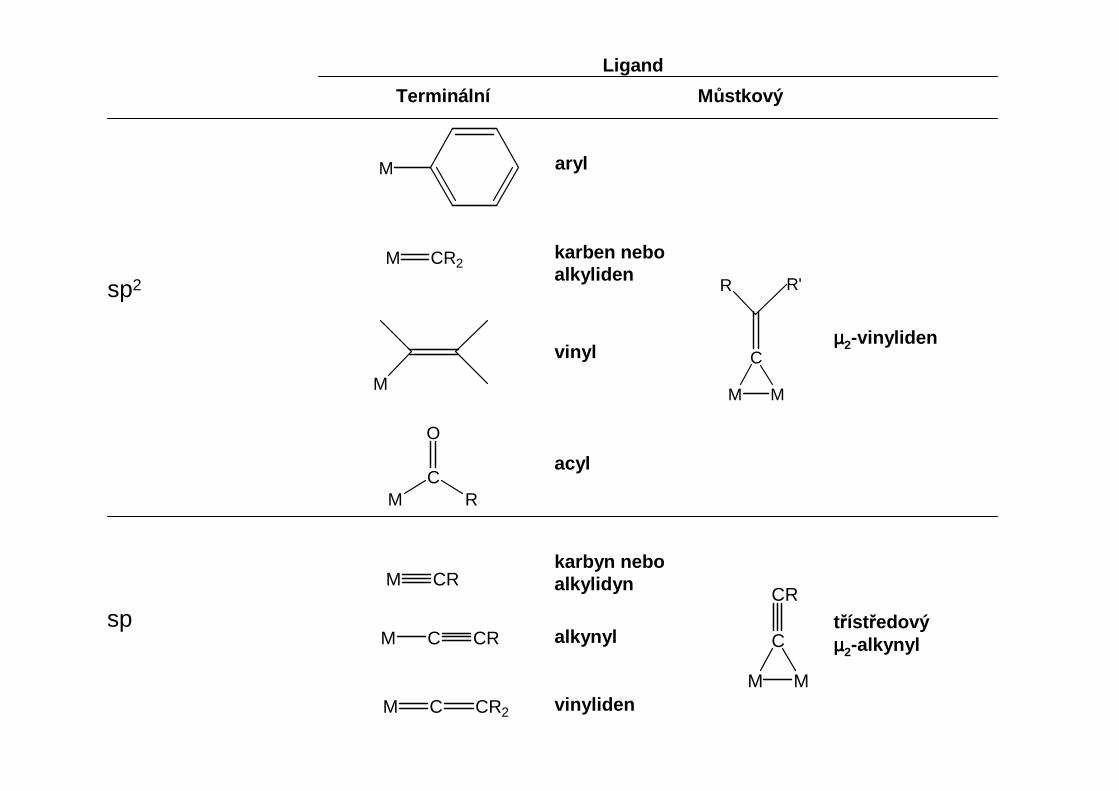
σ-donorové ligandy
Tyto ligandy můžeme klasifikovat podle hybridizace na uhlíku, který je přímo vázán na kov
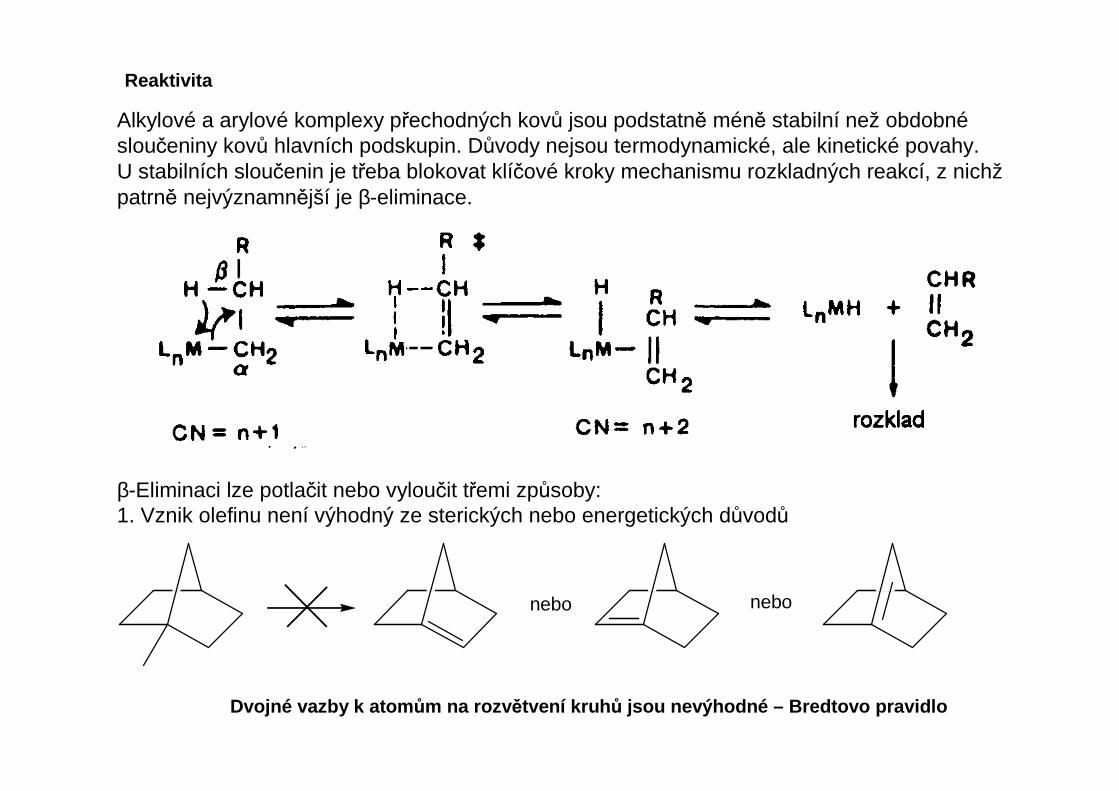
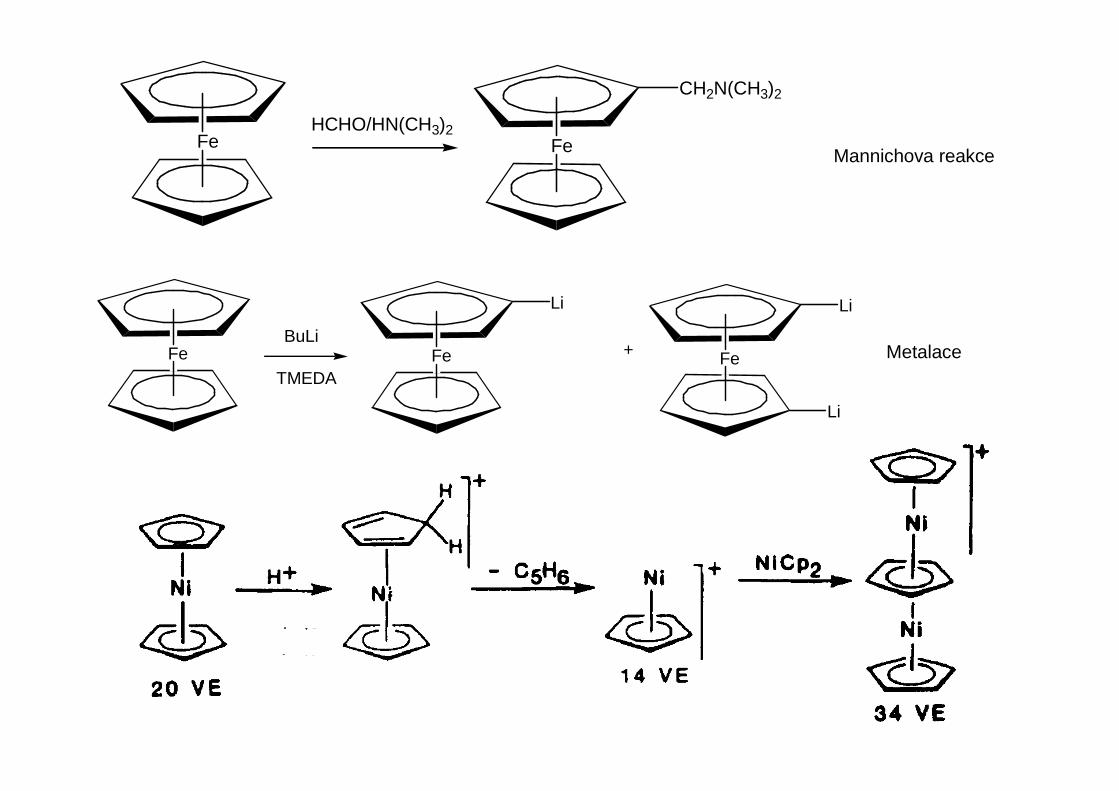
Alkylové a arylové komplexy přechodných kovů jsou podstatně méně stabilní než obdobnésloučeniny kovů hlavních podskupin. Důvody nejsou termodynamické, ale kinetické povahy.U stabilních sloučenin je třeba blokovat klíčové kroky mechanismu rozkladných reakcí, z nichžpatrně nejvýznamnější je β-eliminace.
β-Eliminaci lze potlačit nebo vyloučit třemi způsoby:1. Vznik olefinu není výhodný ze sterických nebo energetických důvodů
nebo nebo
Dvojné vazby k atom ům na rozv ětvení kruh ů jsou nevýhodné – Bredtovo pravidlo
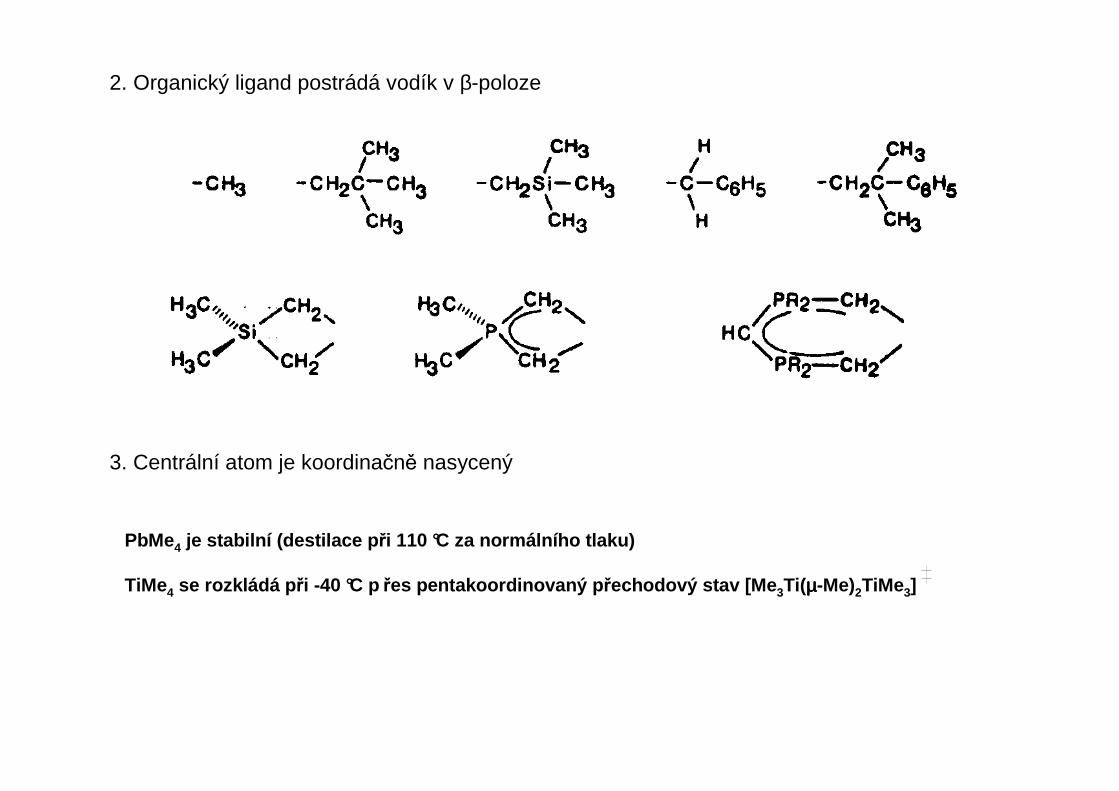
2. Organický ligand postrádá vodík v β-poloze
3. Centrální atom je koordinačně nasycený
PbMe4 je stabilní (destilace p ři 110 °C za normálního tlaku)
TiMe4 se rozkládá p ři -40 °C p řes pentakoordinovaný p řechodový stav [Me 3Ti(µµµµ-Me)2TiMe3]
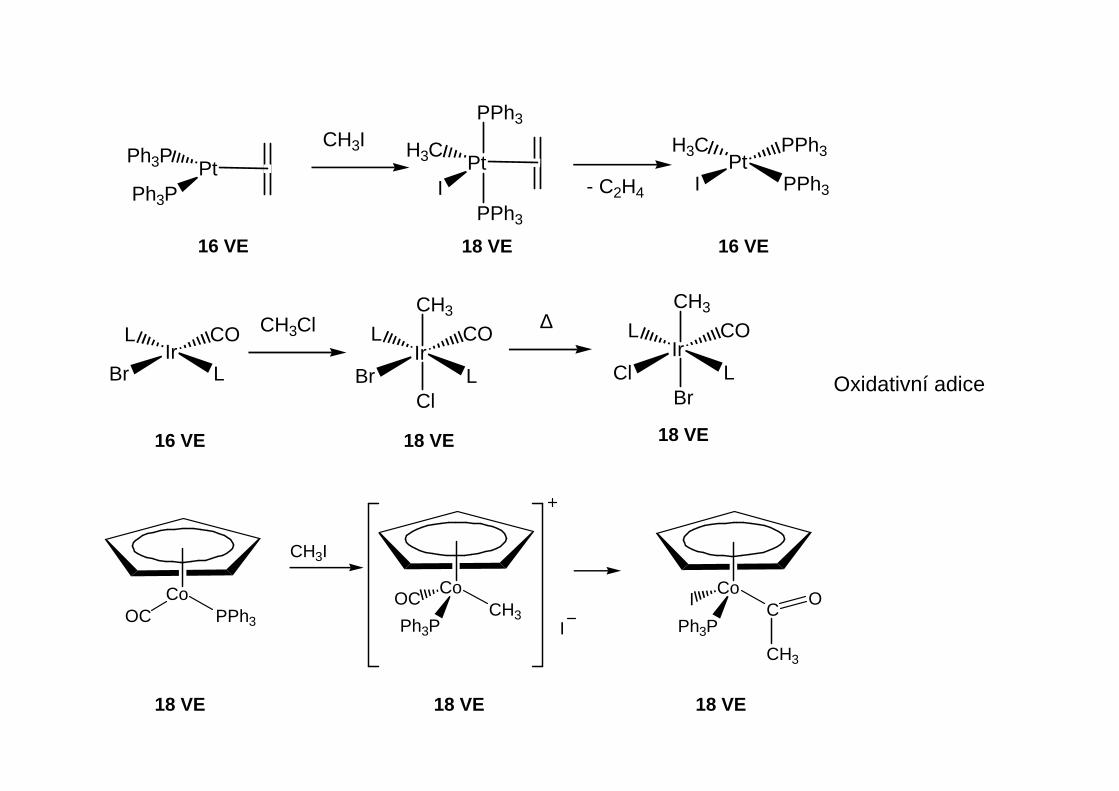
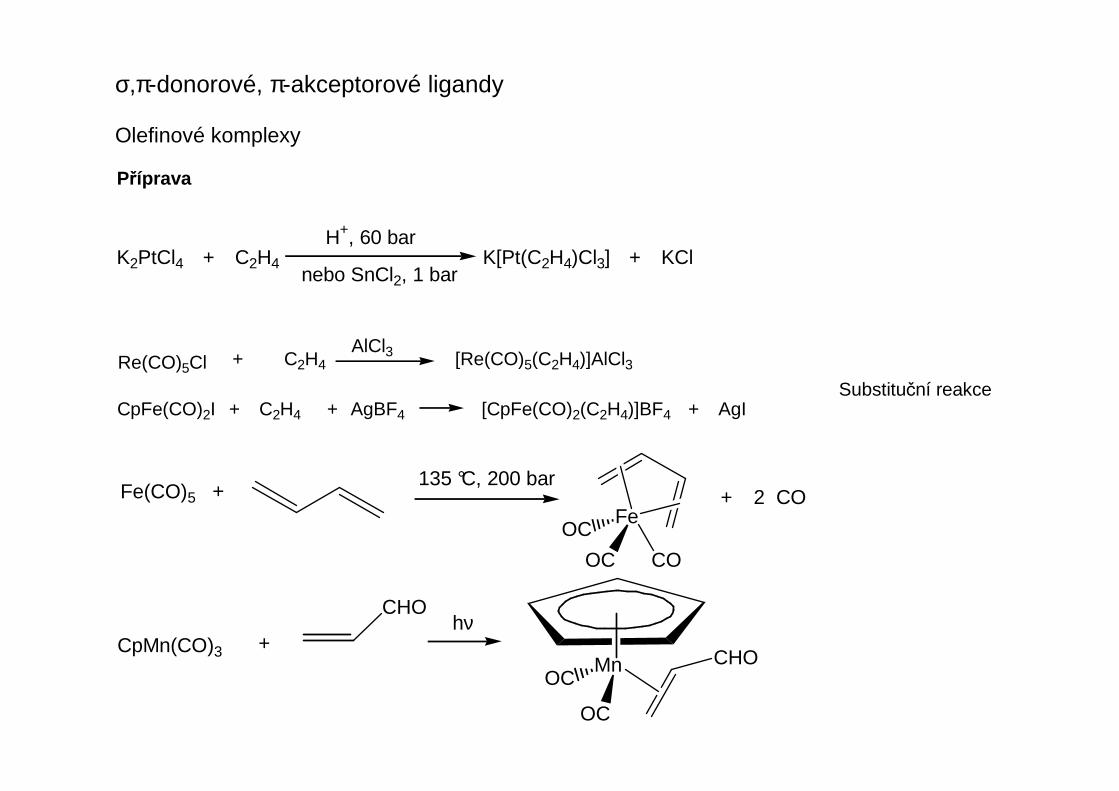
σ-donorové, π-akceptorové ligandy
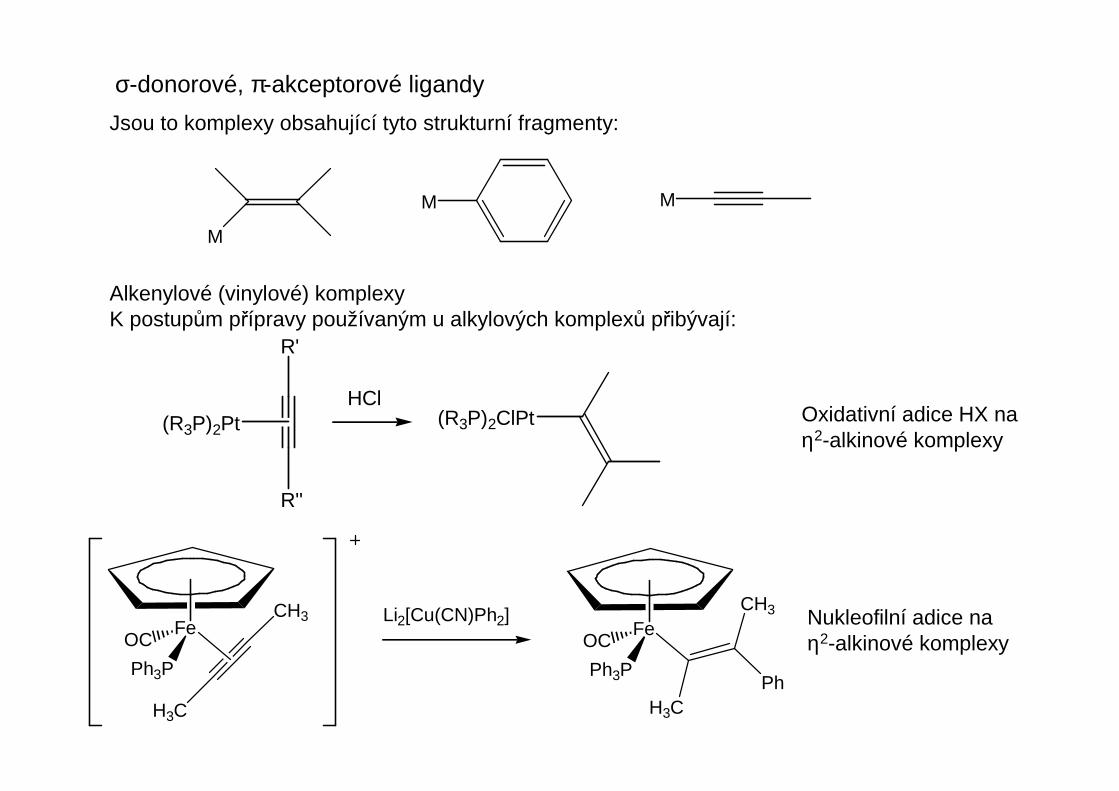
Jsou to komplexy obsahující tyto strukturní fragmenty:
M
MM
Alkenylové (vinylové) komplexyK postupům přípravy používaným u alkylových komplexů přibývají:
Oxidativní adice HX naη2-alkinové komplexy
Nukleofilní adice naη2-alkinové komplexy
R'
R''
(R3P)2PtHCl
(R3P)2ClPt
Fe
Ph3P
OC
CH3
H3C
Li2[Cu(CN)Ph2]Fe
Ph3P
OC
H3C
CH3
Ph
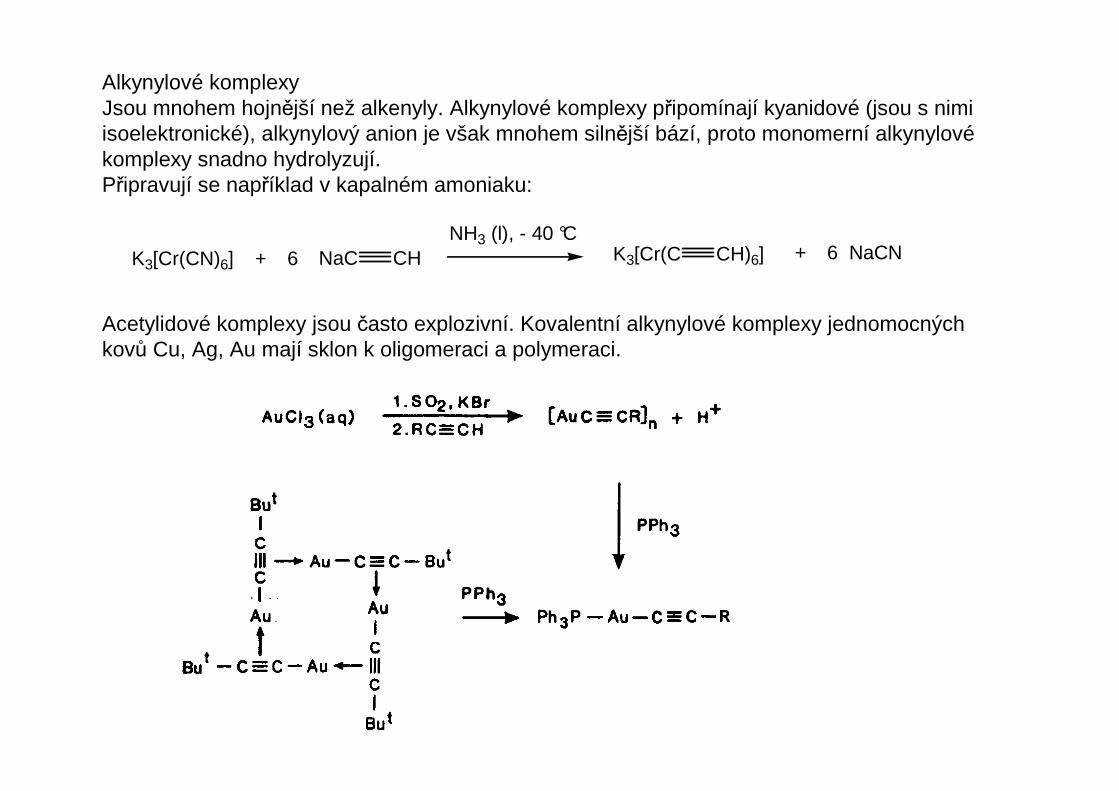
Alkynylové komplexyJsou mnohem hojnější než alkenyly. Alkynylové komplexy připomínají kyanidové (jsou s nimiisoelektronické), alkynylový anion je však mnohem silnější bází, proto monomerní alkynylovékomplexy snadno hydrolyzují.Připravují se například v kapalném amoniaku:
K3[Cr(CN)6] + 6 NaC CHNH3 (l), - 40 °C
K3[Cr(C CH)6] + 6 NaCN
Acetylidové komplexy jsou často explozivní. Kovalentní alkynylové komplexy jednomocnýchkovů Cu, Ag, Au mají sklon k oligomeraci a polymeraci.
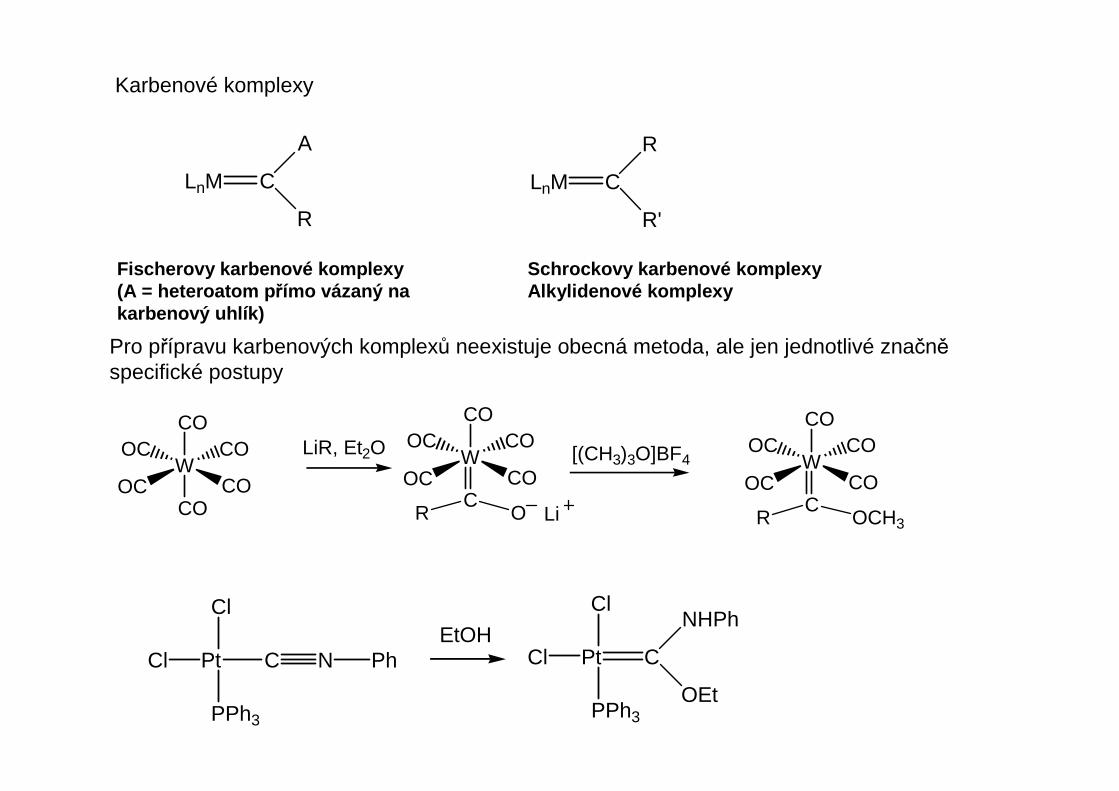
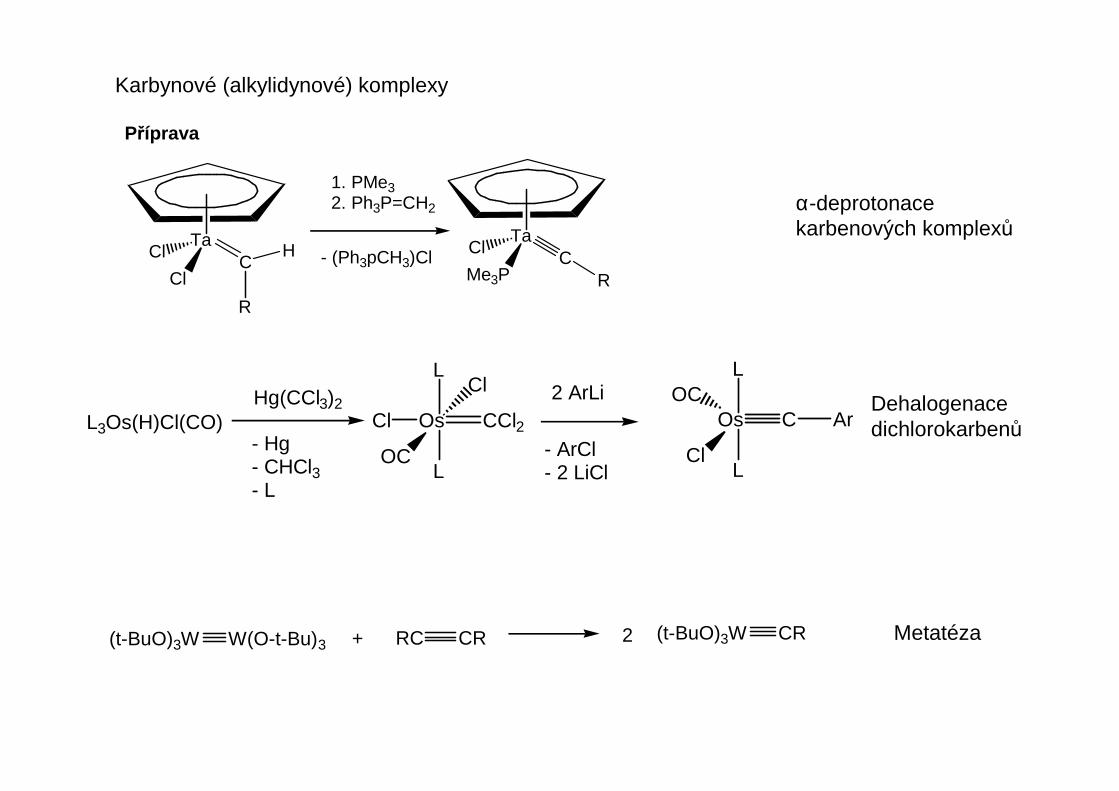
Karbenové komplexy
LnM C
A
R
LnM C
R
R'
Fischerovy karbenové komplexy(A = heteroatom p římo vázaný nakarbenový uhlík)
Pro přípravu karbenových komplexů neexistuje obecná metoda, ale jen jednotlivé značněspecifické postupy
OCW
OC CO
CO
CO
CO
LiR, Et2O
OCW
OC CO
CO
CO
CR O Li
OCW
OC CO
CO
CO
CR OCH3
[(CH3)3O]BF4
Cl
PtCl
PPh3
C N PhEtOH
Cl
PtCl
PPh3
C
NHPh
OEt
N
C
N N
C
N
PhPh
Ph Ph
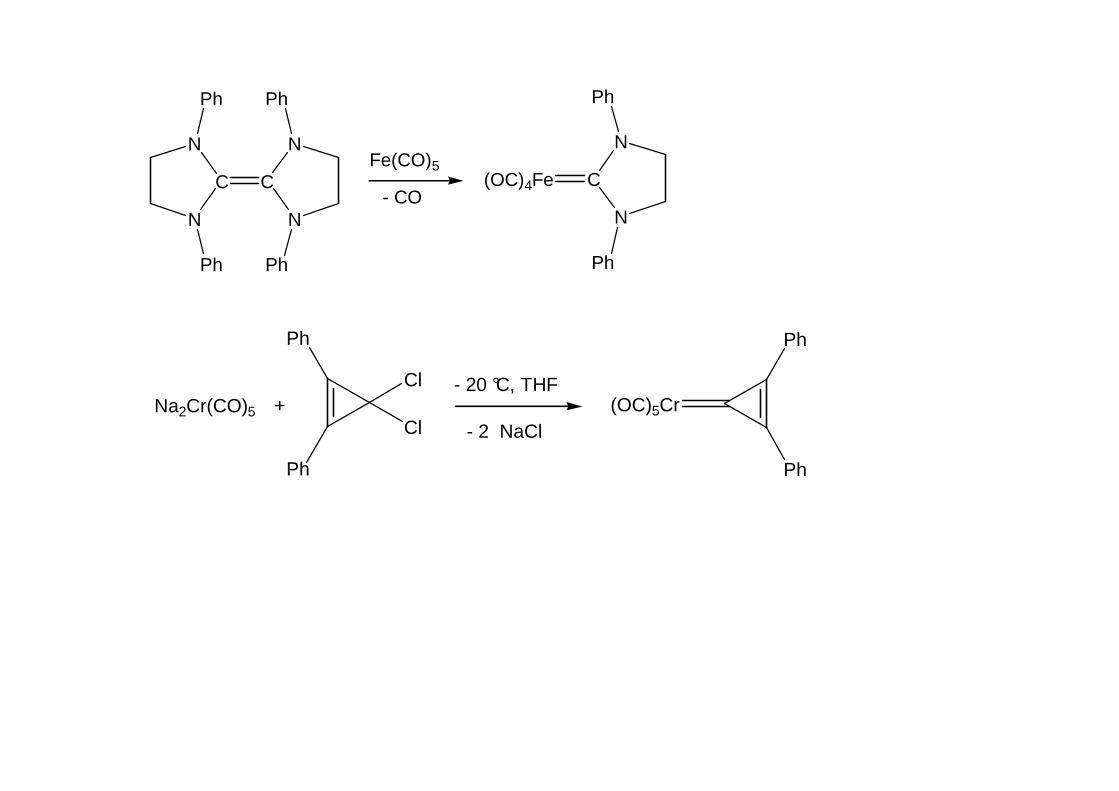
(OC)4Fe
N
C
N
Ph
Ph
Fe(CO)5
- CO
Ph
Ph
Cl
ClNa2Cr(CO)5 +
Ph
Ph
(OC)5Cr- 20 °C, THF
- 2 NaCl
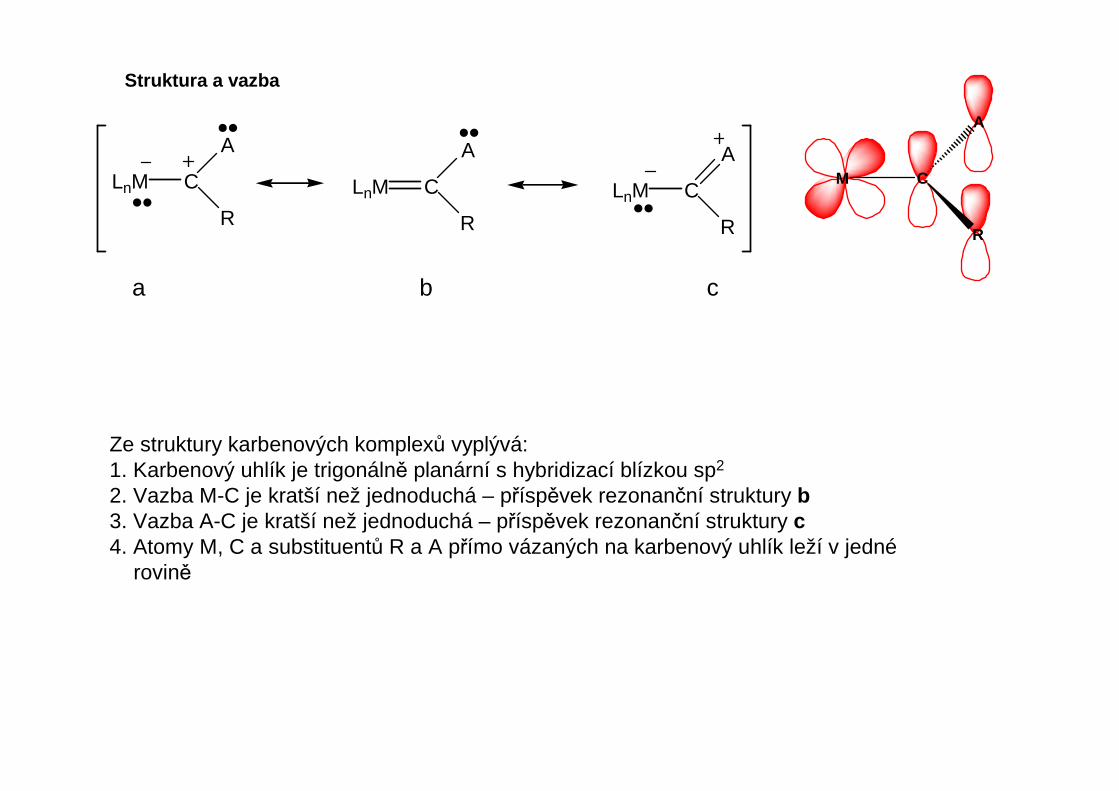
Struktura a vazba
LnM C
A
R
LnM C
A
R
LnM C
A
R
M C
A
R
Ze struktury karbenových komplexů vyplývá:1. Karbenový uhlík je trigonálně planární s hybridizací blízkou sp2
2. Vazba M-C je kratší než jednoduchá – příspěvek rezonanční struktury b3. Vazba A-C je kratší než jednoduchá – příspěvek rezonanční struktury c4. Atomy M, C a substituentů R a A přímo vázaných na karbenový uhlík leží v jedné
rovině
a b c
Reaktivita
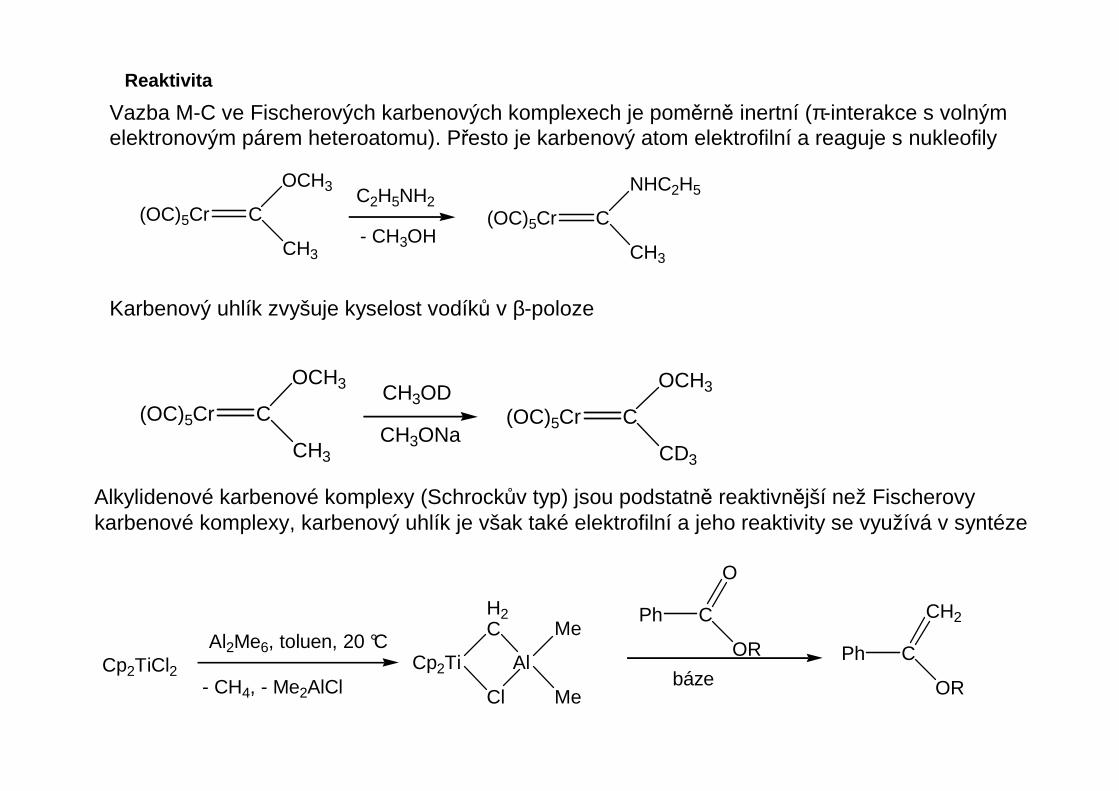
Vazba M-C ve Fischerových karbenových komplexech je poměrně inertní (π-interakce s volnýmelektronovým párem heteroatomu). Přesto je karbenový atom elektrofilní a reaguje s nukleofily
(OC)5Cr C
OCH3
CH3
(OC)5Cr C
NHC2H5
CH3
C2H5NH2
- CH3OH
Karbenový uhlík zvyšuje kyselost vodíků v β-poloze
(OC)5Cr C
OCH3
CH3CH3ONa
(OC)5Cr C
OCH3
CD3
CH3OD
Alkylidenové karbenové komplexy (Schrockův typ) jsou podstatně reaktivnější než Fischerovykarbenové komplexy, karbenový uhlík je však také elektrofilní a jeho reaktivity se využívá v syntéze
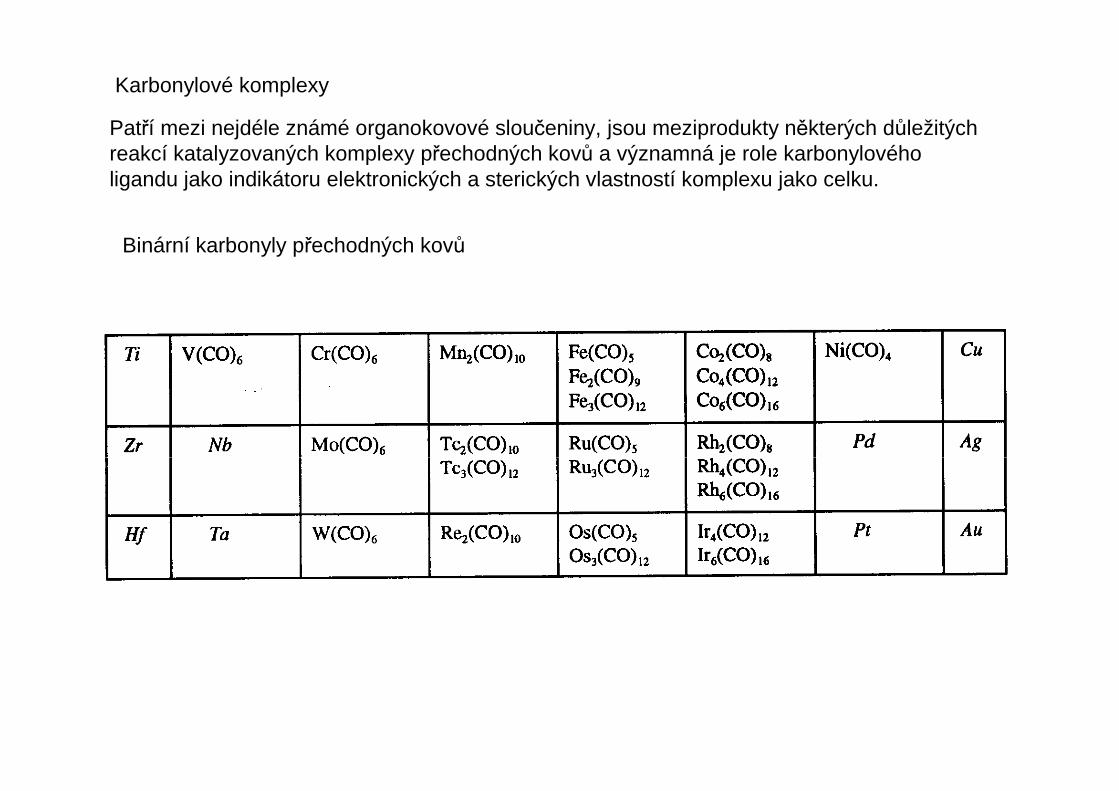
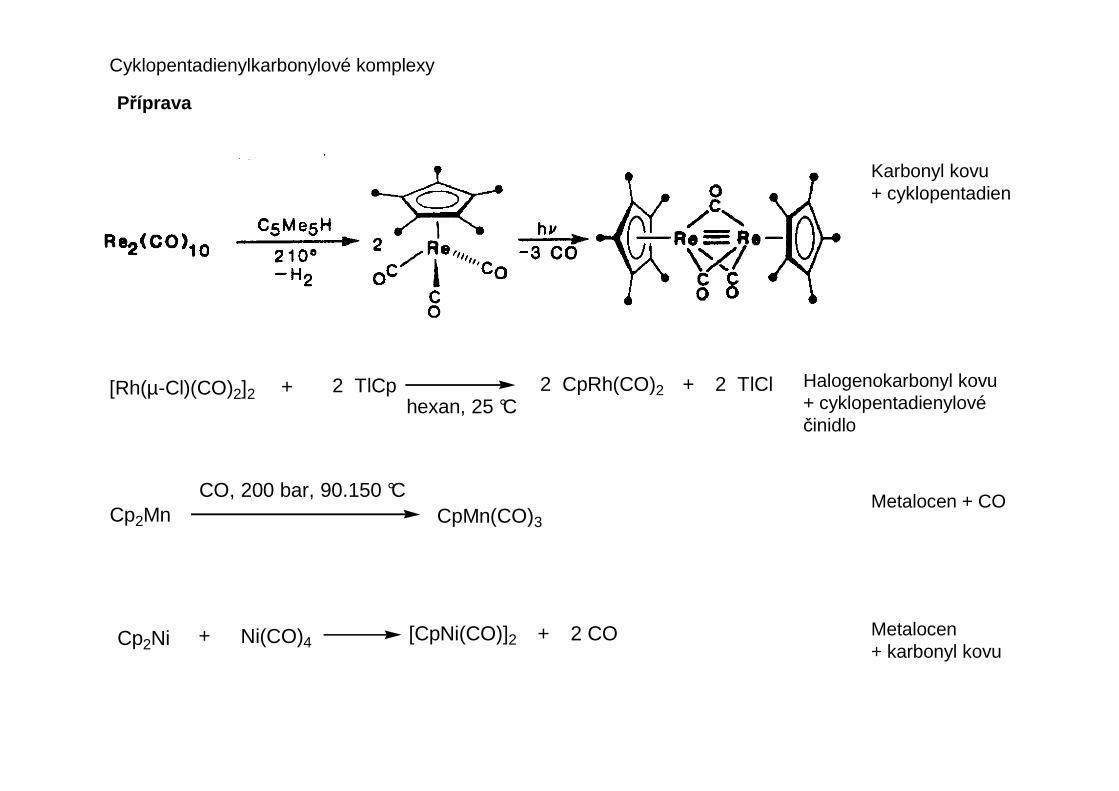
Patří mezi nejdéle známé organokovové sloučeniny, jsou meziprodukty některých důležitýchreakcí katalyzovaných komplexy přechodných kovů a významná je role karbonylovéholigandu jako indikátoru elektronických a sterických vlastností komplexu jako celku.
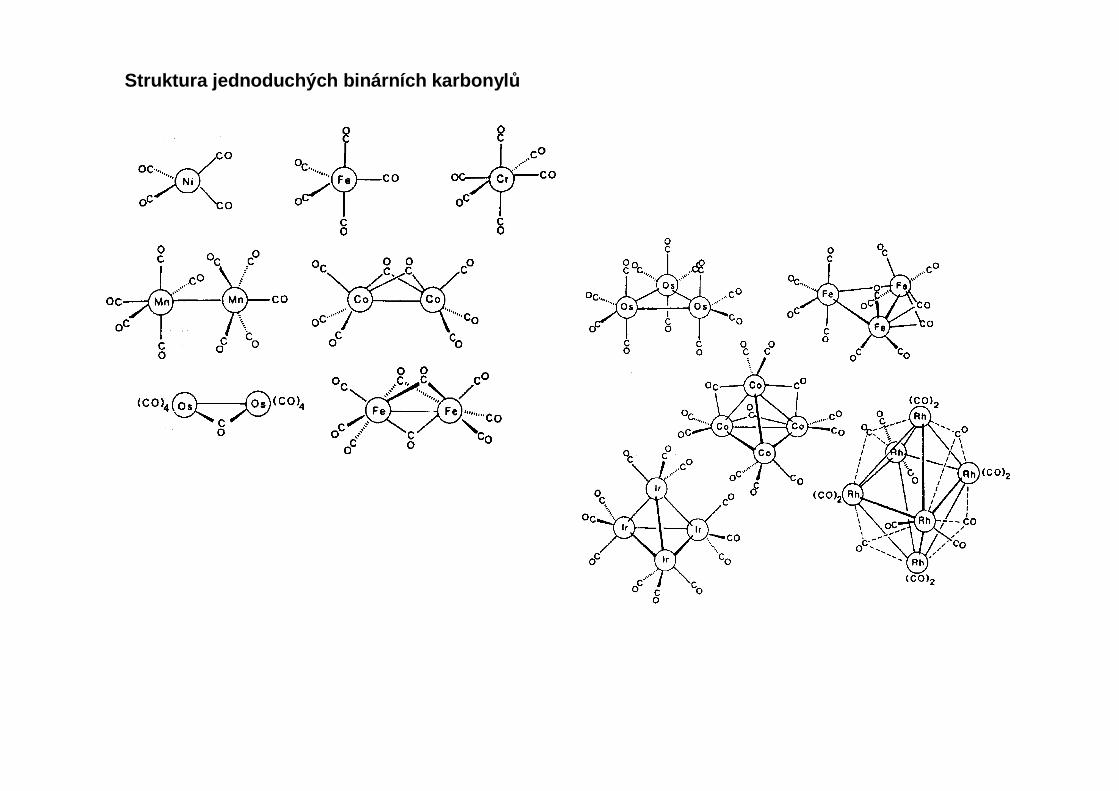
Binární karbonyly přechodných kovů
Struktura jednoduchých binárních karbonyl ů
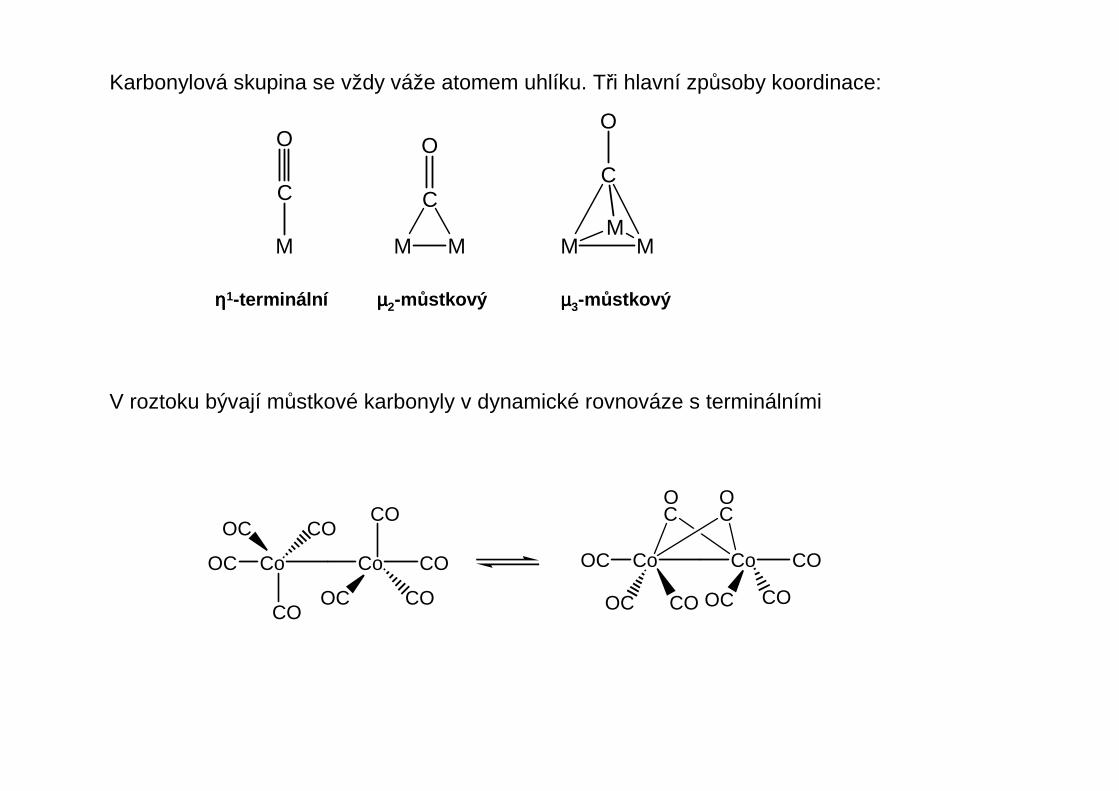
Karbonylová skupina se vždy váže atomem uhlíku. Tři hlavní způsoby koordinace:
M
C
MM
M M
C
O
C
O
M
O
ηηηη1-terminální µµµµ2-můstkový µµµµ3-můstkový
V roztoku bývají můstkové karbonyly v dynamické rovnováze s terminálními
OC Co
CO
Co CO
COOC
COCO
OC
OC Co Co CO
COOCCOOC
OC
OC
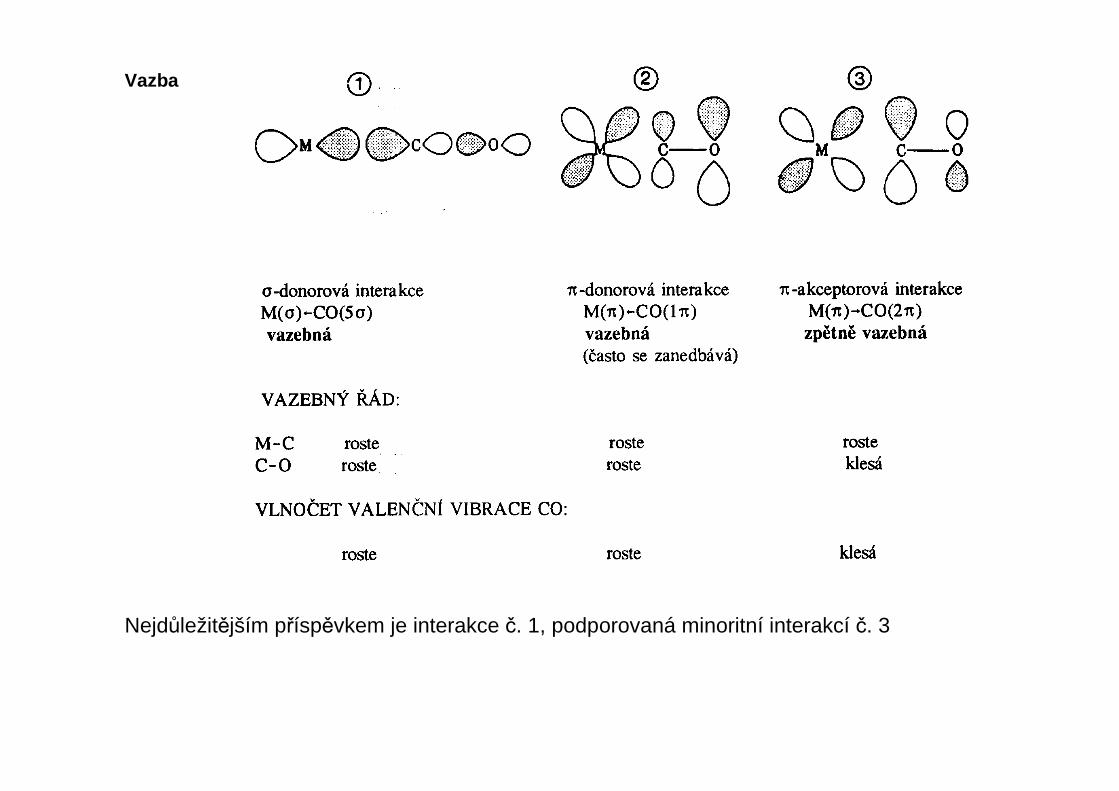
Vazba
Nejdůležitějším příspěvkem je interakce č. 1, podporovaná minoritní interakcí č. 3
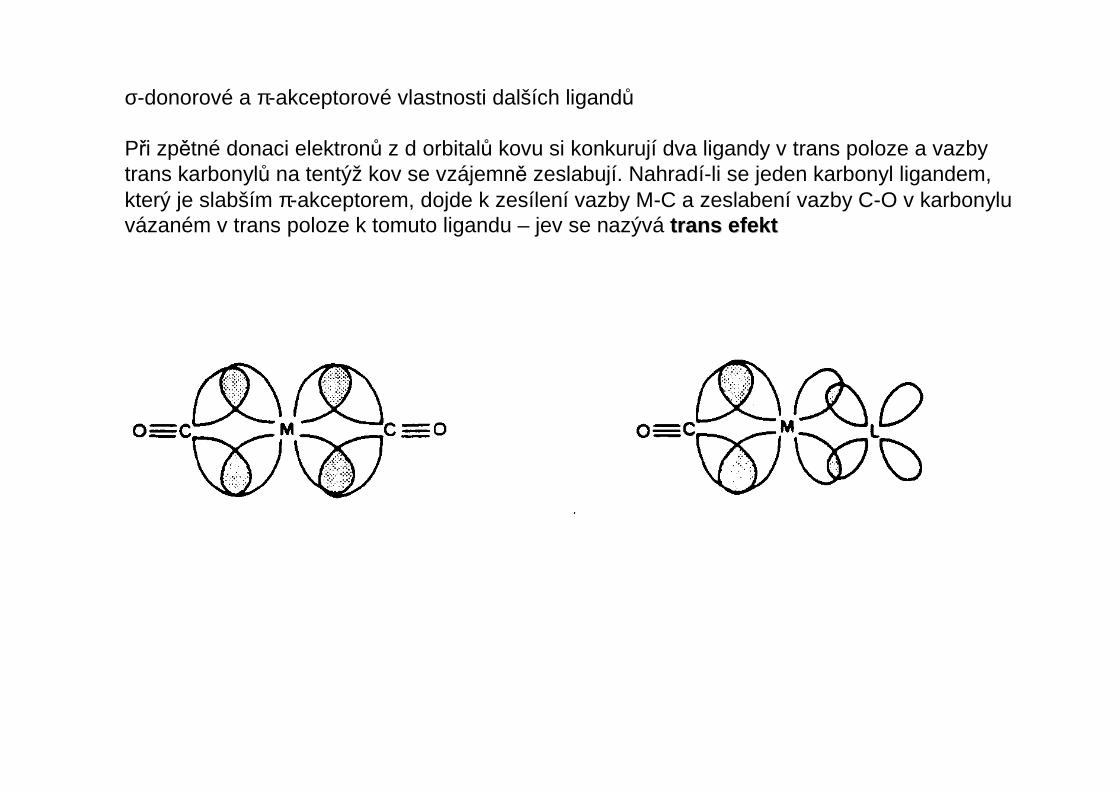
σ-donorové a π-akceptorové vlastnosti dalších ligandů
Při zpětné donaci elektronů z d orbitalů kovu si konkurují dva ligandy v trans poloze a vazbytrans karbonylů na tentýž kov se vzájemně zeslabují. Nahradí-li se jeden karbonyl ligandem,který je slabším π-akceptorem, dojde k zesílení vazby M-C a zeslabení vazby C-O v karbonyluvázaném v trans poloze k tomuto ligandu – jev se nazývá trans efekttrans efekt
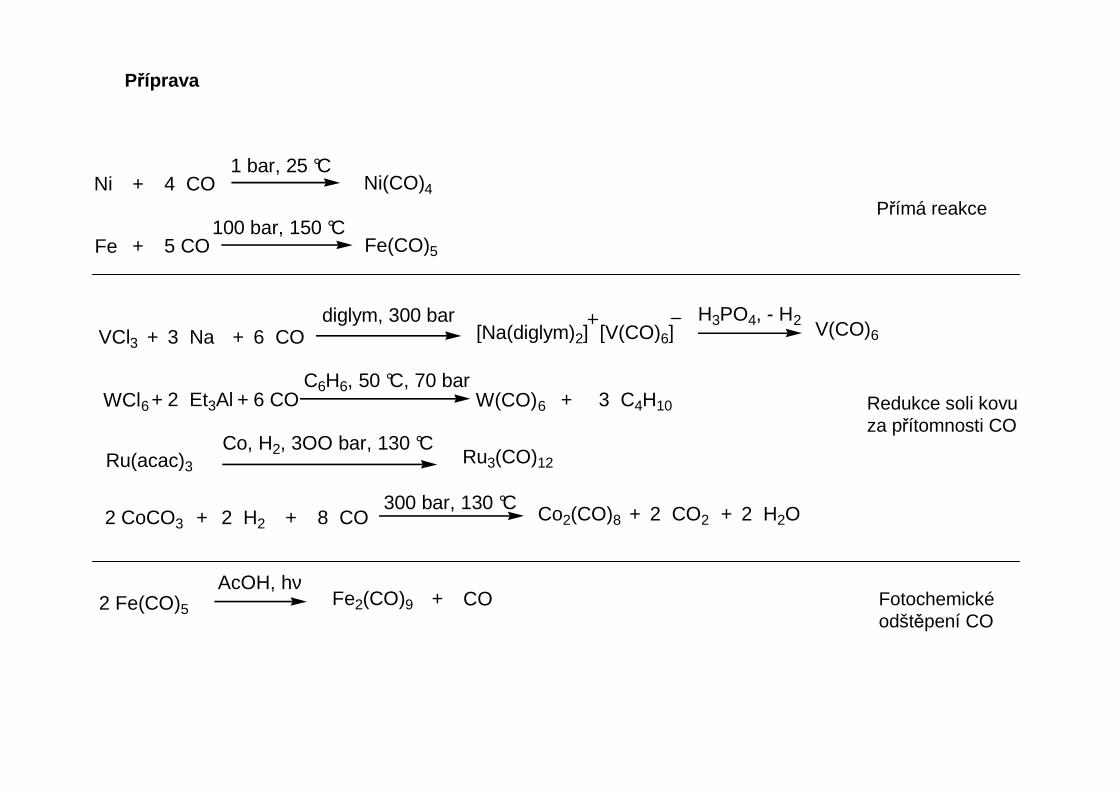
Příprava
Ni + 4 CO Ni(CO)41 bar, 25 °C
Fe + 5 CO Fe(CO)5
100 bar, 150 °C
VCl3 + 3 Na + 6 COdiglym, 300 bar
[Na(diglym)2] [V(CO)6]H3PO4, - H2 V(CO)6
WCl6 + 2 Et3Al + 6 CO W(CO)6 + 3 C4H10
C6H6, 50 °C, 70 bar
Ru(acac)3
Co, H2, 3OO bar, 130 °CRu3(CO)12
2 CoCO3 + 2 H2 + 8 CO300 bar, 130 °C
Co2(CO)8 + 2 CO2 + 2 H2O
2 Fe(CO)5
AcOH, hνFe2(CO)9 + CO
Přímá reakce
Redukce soli kovuza přítomnosti CO
Fotochemickéodštěpení CO
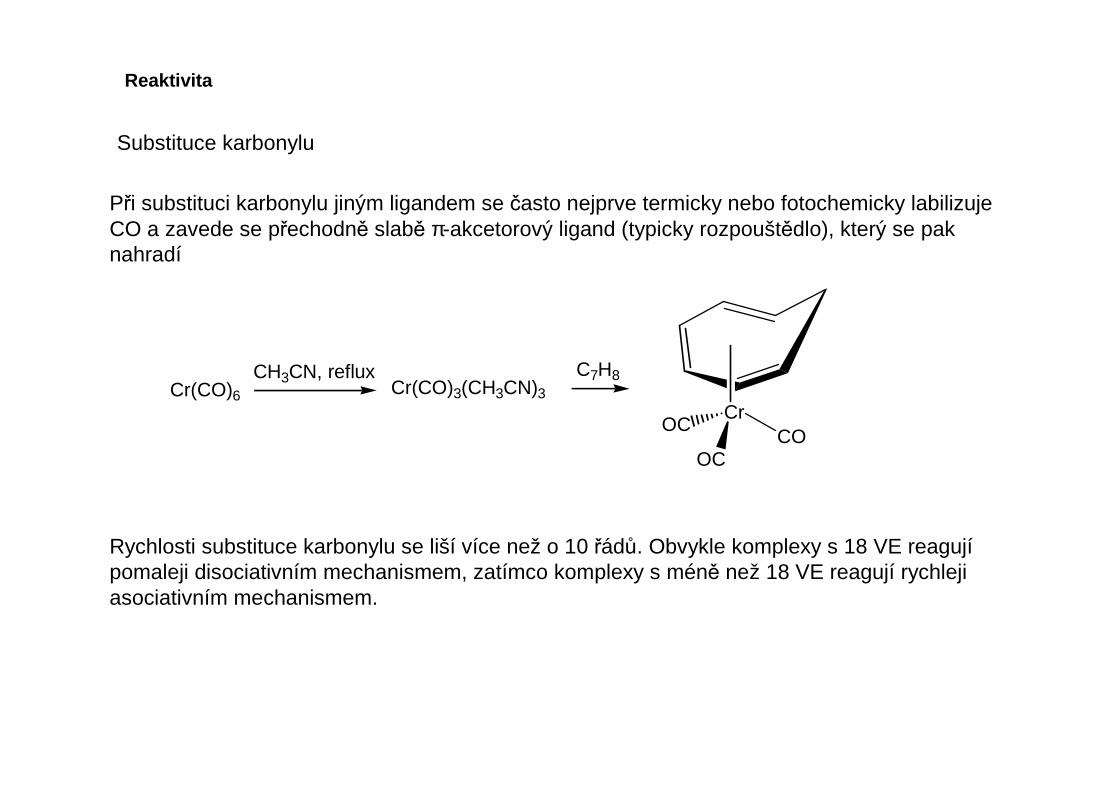
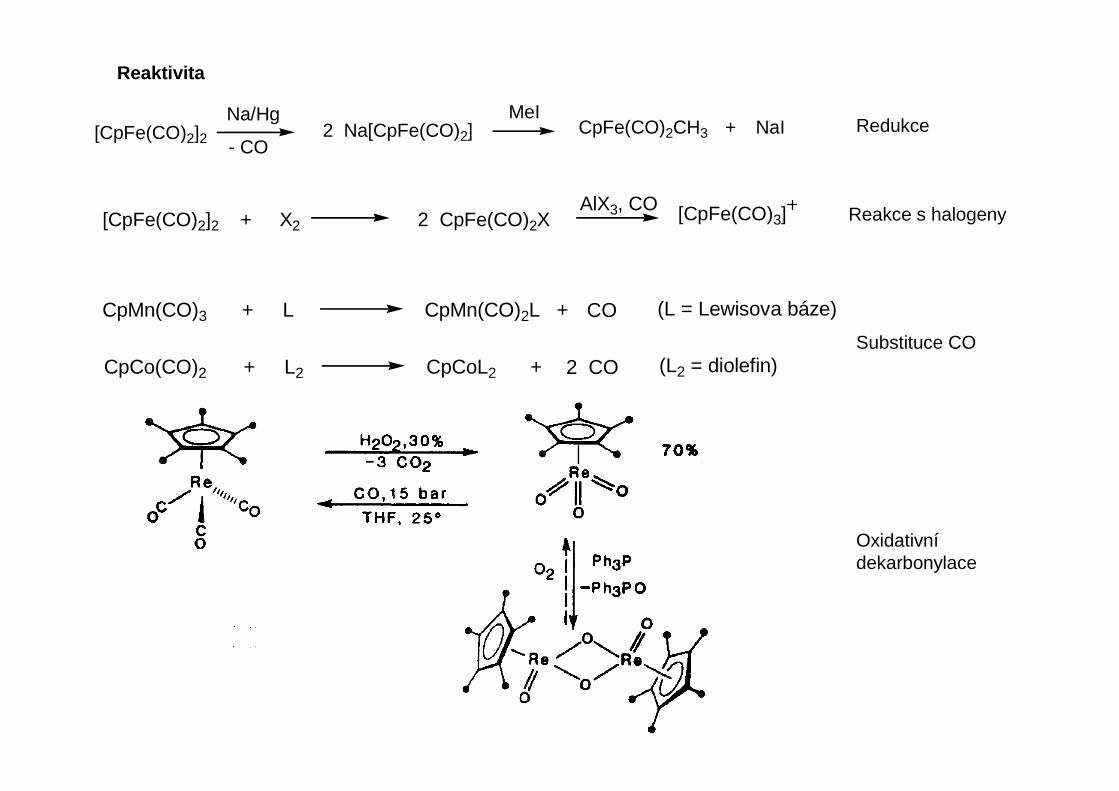
Reaktivita
Při substituci karbonylu jiným ligandem se často nejprve termicky nebo fotochemicky labilizuje CO a zavede se přechodně slabě π-akcetorový ligand (typicky rozpouštědlo), který se pak nahradí
CH3CN, refluxCr(CO)6 Cr(CO)3(CH3CN)3
C7H8
CrCO
OC
OC
Rychlosti substituce karbonylu se liší více než o 10 řádů. Obvykle komplexy s 18 VE reagujípomaleji disociativním mechanismem, zatímco komplexy s méně než 18 VE reagují rychlejiasociativním mechanismem.
Substituce karbonylu
Adice nukleofilních činidel na karbonyl
Fe(CO)5 + Na [HB(OMe)3] Na [(CO)4Fe-C(H)=O] + B(OMe)3
Syntéza karbenových komplexů – viz výše
Vznik karbonylátových iontů – viz níže
Disproporcionace
3 Mn2(CO)10 + 12 C6H5N120 °C, - 10 CO 2 [Mn(py)6]2+ + 4 [Mn(CO)5]
Oxidativní dekarbonylace – viz níže vznik halogenokarbonylů
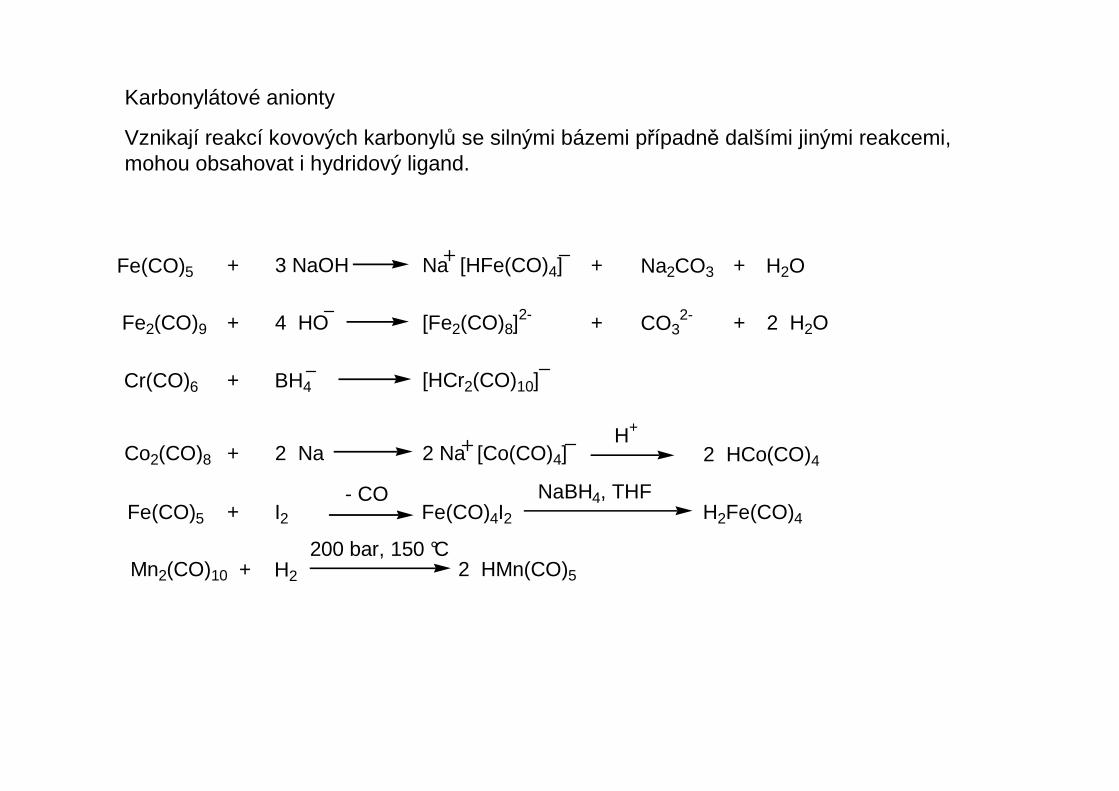
Karbonylátové anionty
Vznikají reakcí kovových karbonylů se silnými bázemi případně dalšími jinými reakcemi, mohou obsahovat i hydridový ligand.
Fe(CO)5 + 3 NaOH Na [HFe(CO)4] + Na2CO3 + H2O
Fe2(CO)9 + 4 HO [Fe2(CO)8]2- + CO3
2- + 2 H2O
Cr(CO)6 + BH4 [HCr2(CO)10]
Co2(CO)8 + 2 Na 2 Na [Co(CO)4]H+
2 HCo(CO)4
Fe(CO)5 + I2 Fe(CO)4I2 H2Fe(CO)4- CO NaBH4, THF
Mn2(CO)10 + H2 2 HMn(CO)5
200 bar, 150 °C
Název „hydridy“ je založen na formálním oxidačním stupni, což nemusí odpovídat chemickéreaktivitě. Vodík na přechodném kovu se může projevovat jako hydrid i jako proton (kyselostHCo(CO)4, pKa = 1, je srovnatelná s H2SO4)
CpFe(CO)2H + HCl CpFe(CO)2Cl + H2
HCo(CO)4 + H2O H3O + [Co(CO)4]
Identifikace hydridového ligandu je velmi dobře možná z NMR spektra 1H: hydridy se vyznačujívelkým chemickým posunem k negativním hodnotám (0 až -50 ppm)
Významnými reakcemi karbonylátových iontů jsou alkylace a silylace
[Mn(CO)5] + MeI MeMn(CO)5 + I
[CpW(CO)3] + Me3SiCl CpW(CO)3SiMe3 + Cl
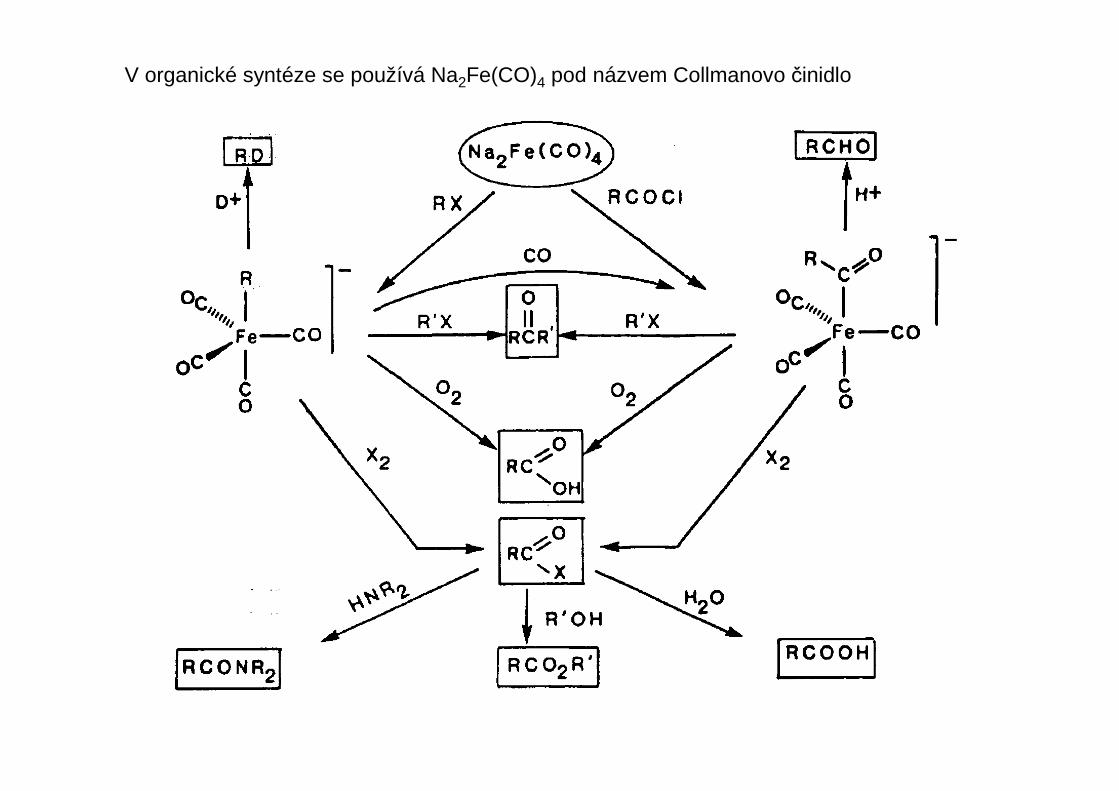
V organické syntéze se používá Na2Fe(CO)4 pod názvem Collmanovo činidlo
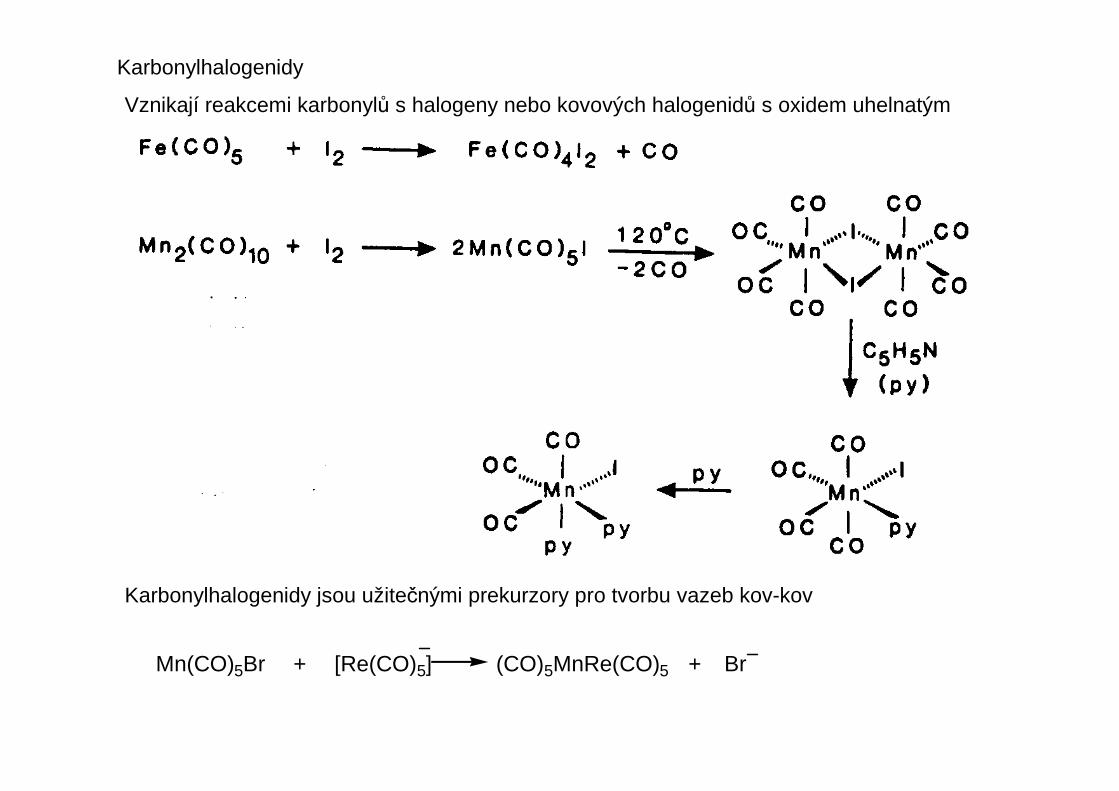
Karbonylhalogenidy
Vznikají reakcemi karbonylů s halogeny nebo kovových halogenidů s oxidem uhelnatým
Karbonylhalogenidy jsou užitečnými prekurzory pro tvorbu vazeb kov-kov
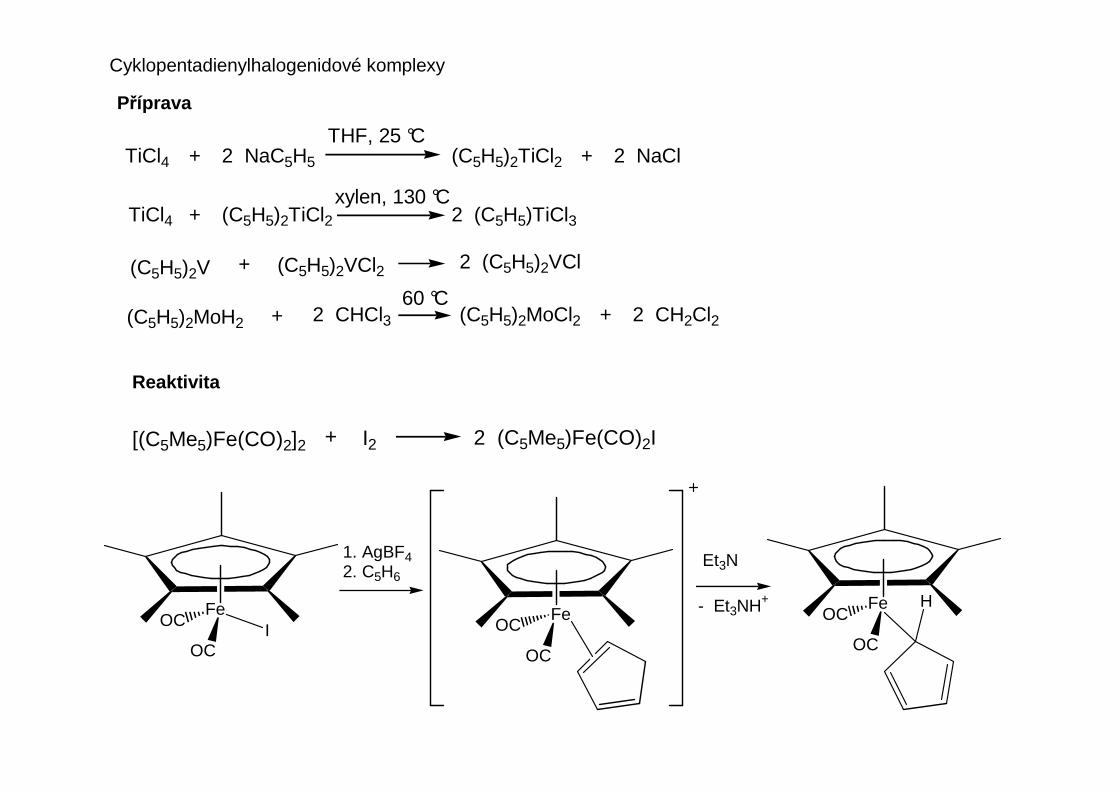
CpFe(CO)2I + C2H4 + AgBF4 [CpFe(CO)2(C2H4)]BF4 + AgISubstituční reakce
Fe(CO)5 +135 °C, 200 bar
Fe
COOC
OC
+ 2 CO
CpMn(CO)3 +
CHOhν
Mn
OC
OCCHO
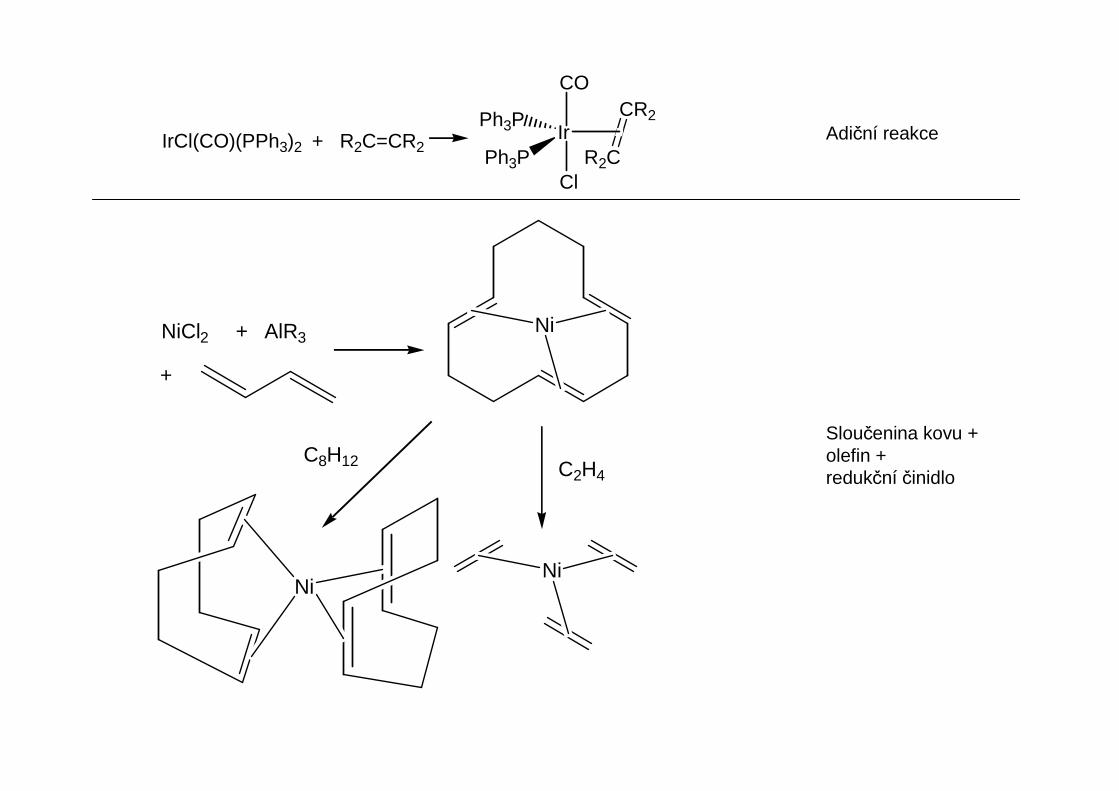
Adiční reakceIrCl(CO)(PPh3)2 + R2C=CR2Ir
Ph3P
Ph3P
CO
Cl
CR2
R2C
Sloučenina kovu +olefin +redukční činidlo
NiNiCl2 + AlR3
+
C2H4C8H12
NiNi
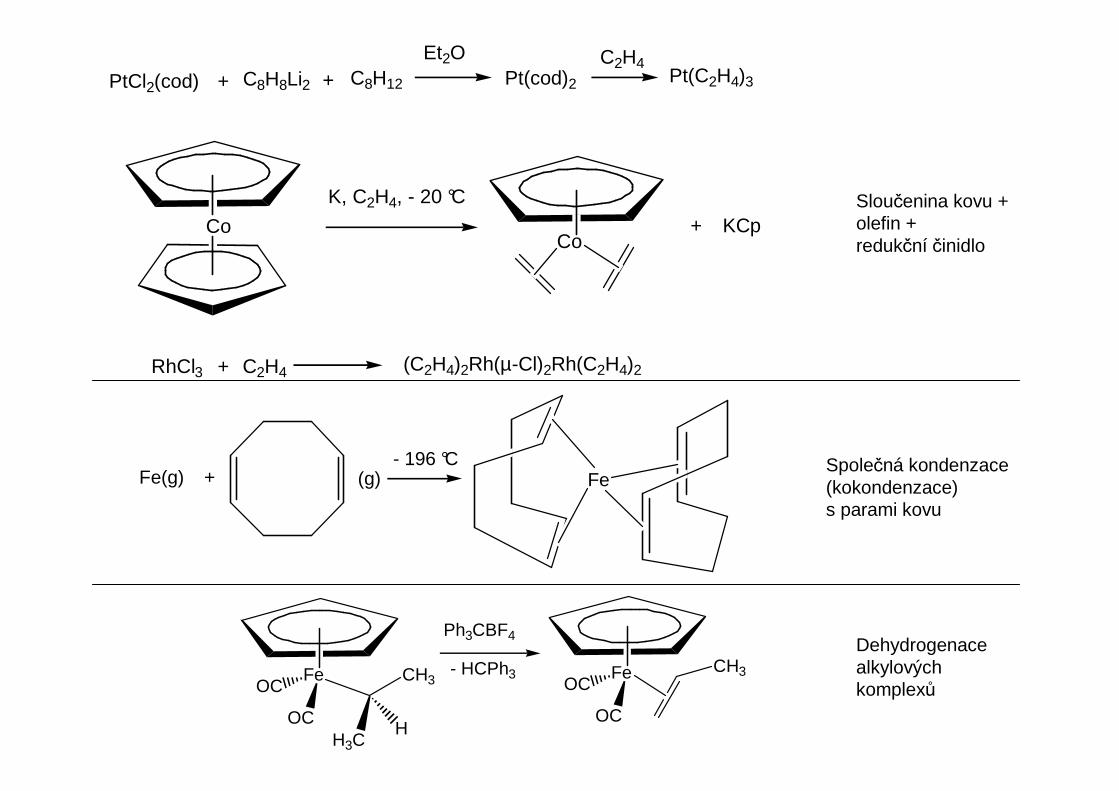
PtCl2(cod) + + C8H12C8H8Li2
Et2OPt(cod)2
C2H4Pt(C2H4)3
CoCo
K, C2H4, - 20 °C
+ KCp
RhCl3 + C2H4 (C2H4)2Rh(µ-Cl)2Rh(C2H4)2
Sloučenina kovu +olefin +redukční činidlo
Fe
OC
OCCH3Fe
OC
OCCH3
H3CH
Ph3CBF4
- HCPh3
Společná kondenzace(kokondenzace)s parami kovu
Dehydrogenacealkylovýchkomplexů
FeFe(g) + (g)- 196 °C
Struktura a vazba
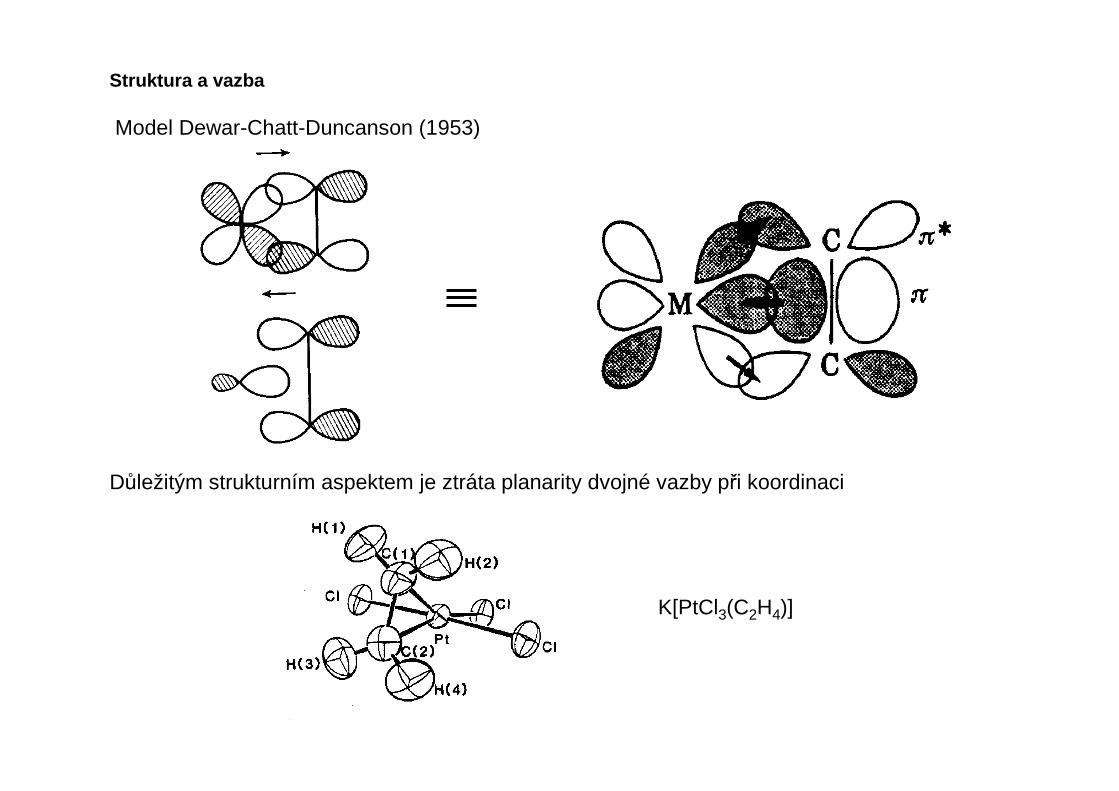
Model Dewar-Chatt-Duncanson (1953)
Důležitým strukturním aspektem je ztráta planarity dvojné vazby při koordinaci
K[PtCl3(C2H4)]
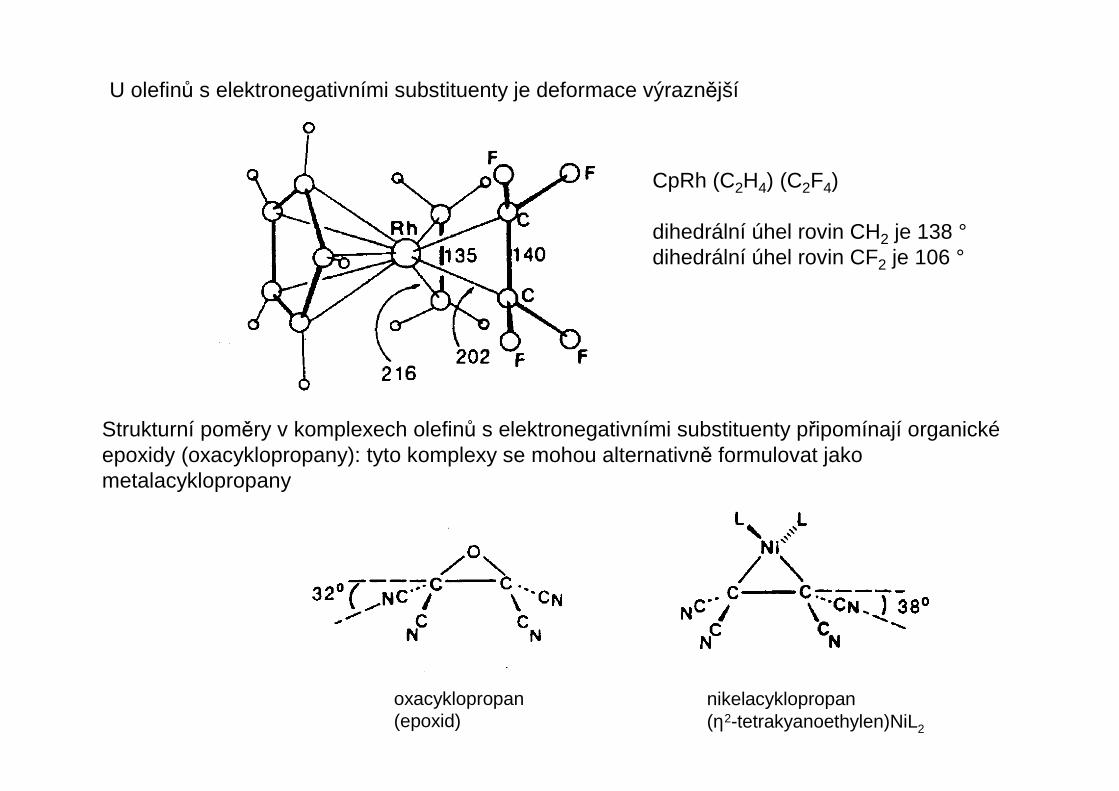
U olefinů s elektronegativními substituenty je deformace výraznější
Strukturní poměry v komplexech olefinů s elektronegativními substituenty připomínají organickéepoxidy (oxacyklopropany): tyto komplexy se mohou alternativně formulovat jako metalacyklopropany
CpRh (C2H4) (C2F4)
dihedrální úhel rovin CH2 je 138 °dihedrální úhel rovin CF2 je 106 °
oxacyklopropan(epoxid)
nikelacyklopropan(η2-tetrakyanoethylen)NiL2
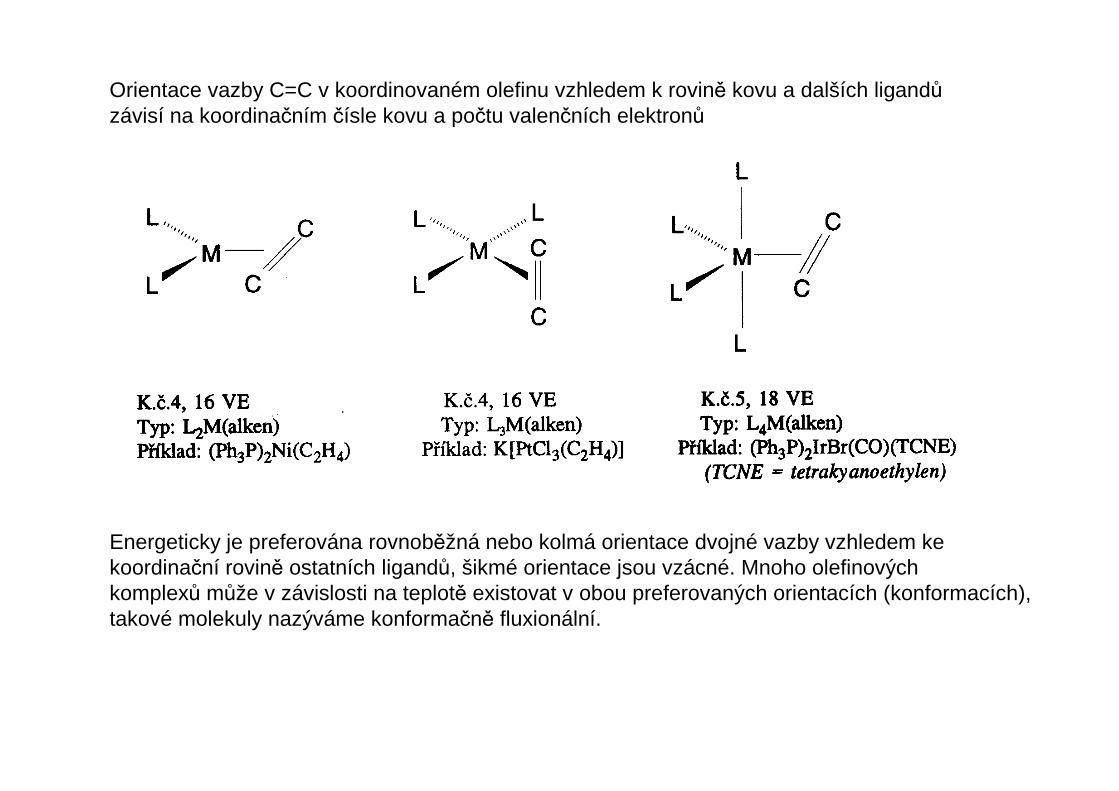
Orientace vazby C=C v koordinovaném olefinu vzhledem k rovině kovu a dalších ligandůzávisí na koordinačním čísle kovu a počtu valenčních elektronů
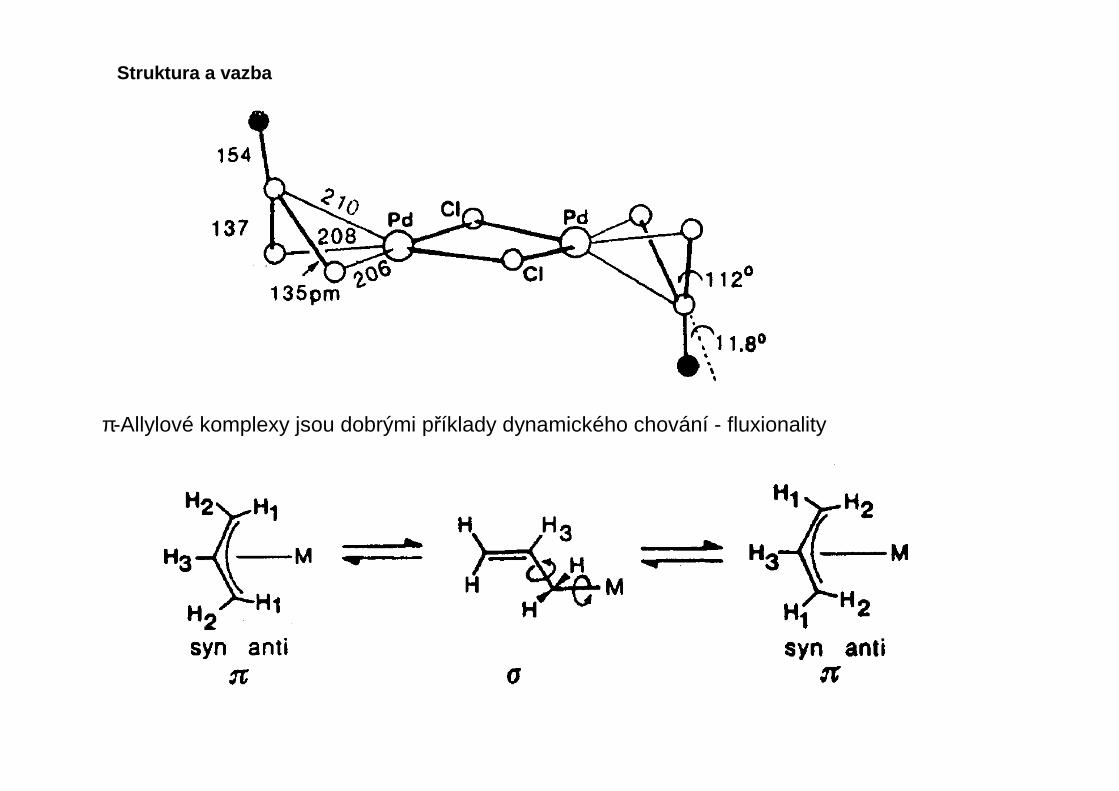
Energeticky je preferována rovnoběžná nebo kolmá orientace dvojné vazby vzhledem kekoordinační rovině ostatních ligandů, šikmé orientace jsou vzácné. Mnoho olefinovýchkomplexů může v závislosti na teplotě existovat v obou preferovaných orientacích (konformacích),takové molekuly nazýváme konformačně fluxionální.
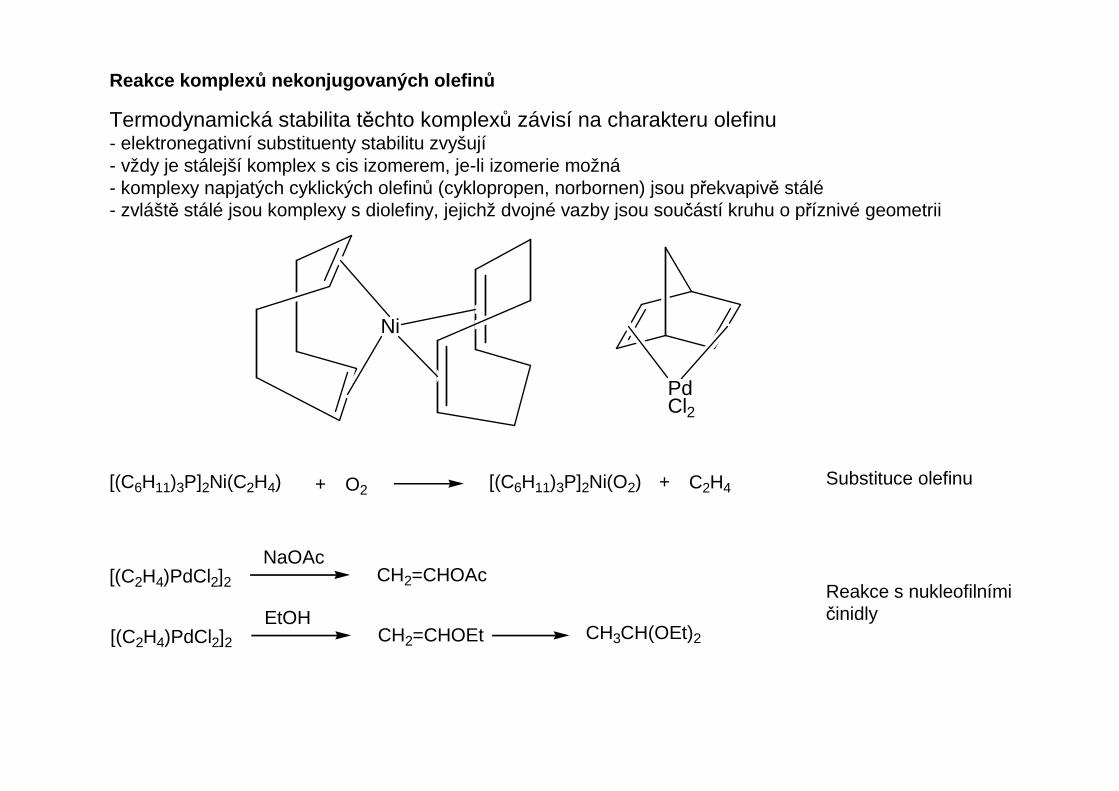
Reakce komplex ů nekonjugovaných olefin ů
Termodynamická stabilita těchto komplexů závisí na charakteru olefinu- elektronegativní substituenty stabilitu zvyšují- vždy je stálejší komplex s cis izomerem, je-li izomerie možná- komplexy napjatých cyklických olefinů (cyklopropen, norbornen) jsou překvapivě stálé- zvláště stálé jsou komplexy s diolefiny, jejichž dvojné vazby jsou součástí kruhu o příznivé geometrii
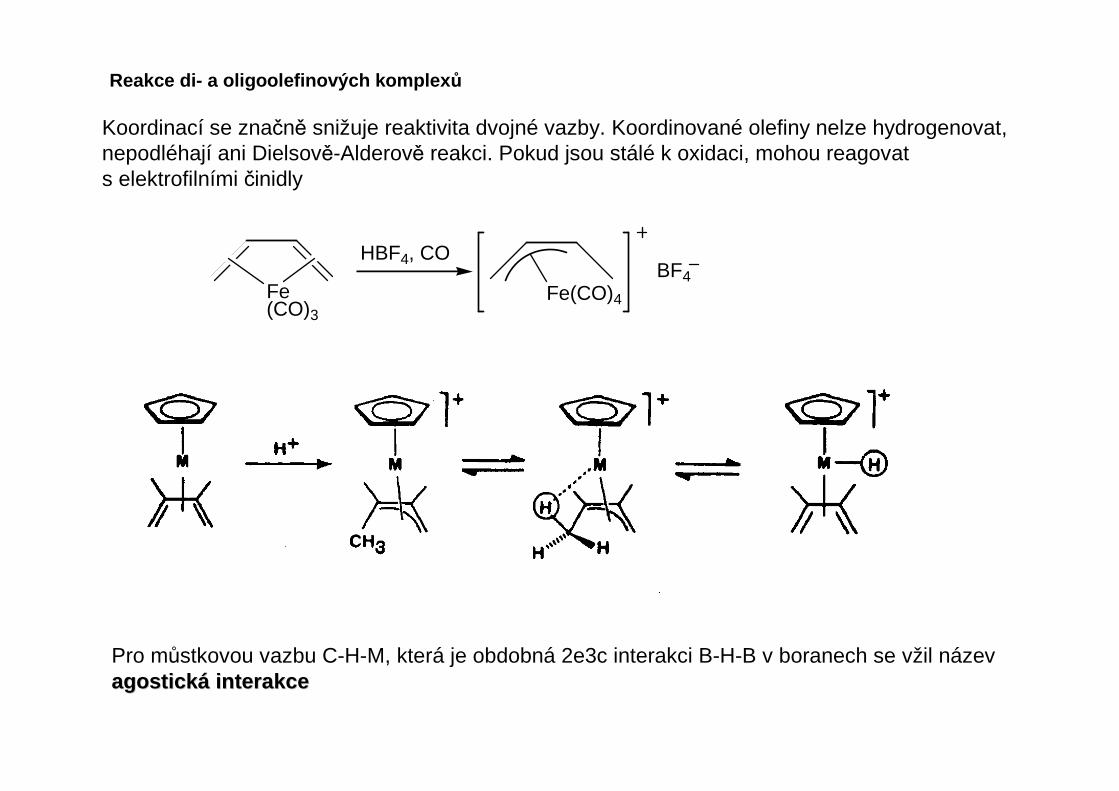
Koordinací se značně snižuje reaktivita dvojné vazby. Koordinované olefiny nelze hydrogenovat,nepodléhají ani Dielsově-Alderově reakci. Pokud jsou stálé k oxidaci, mohou reagovats elektrofilními činidly
Fe(CO)3
Fe(CO)4
HBF4, COBF4
Pro můstkovou vazbu C-H-M, která je obdobná 2e3c interakci B-H-B v boranech se vžil názevagostickagostick áá interakceinterakce
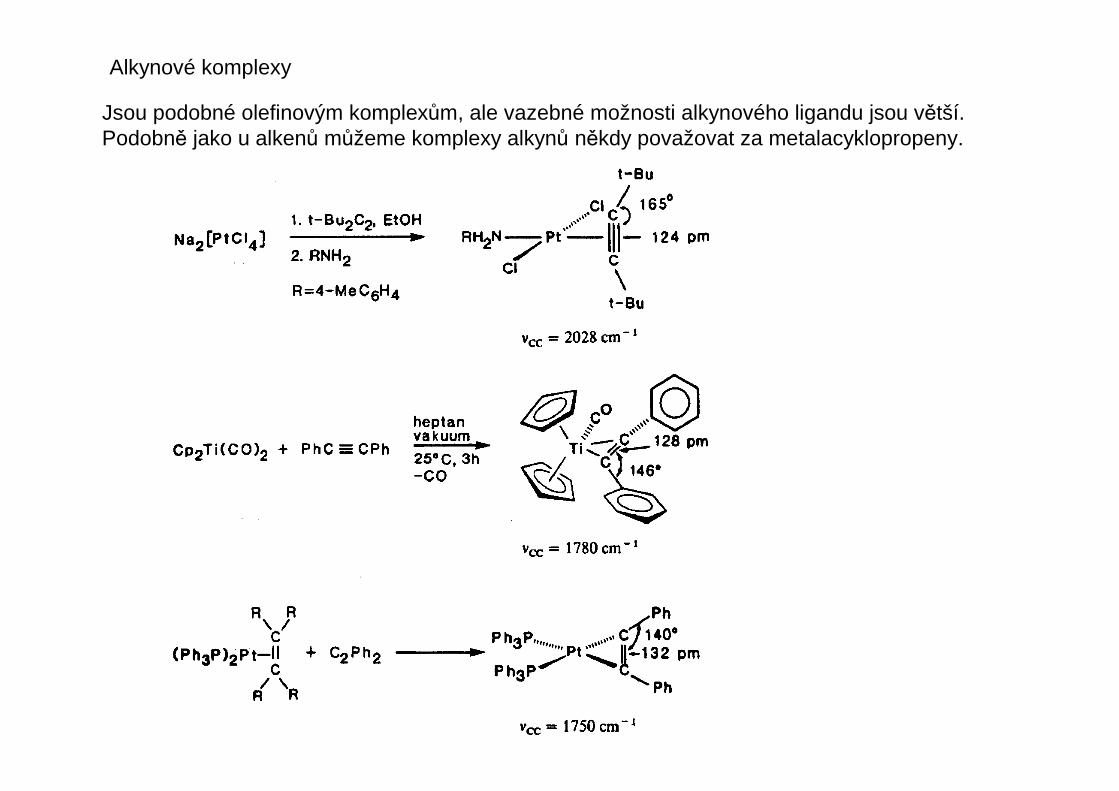
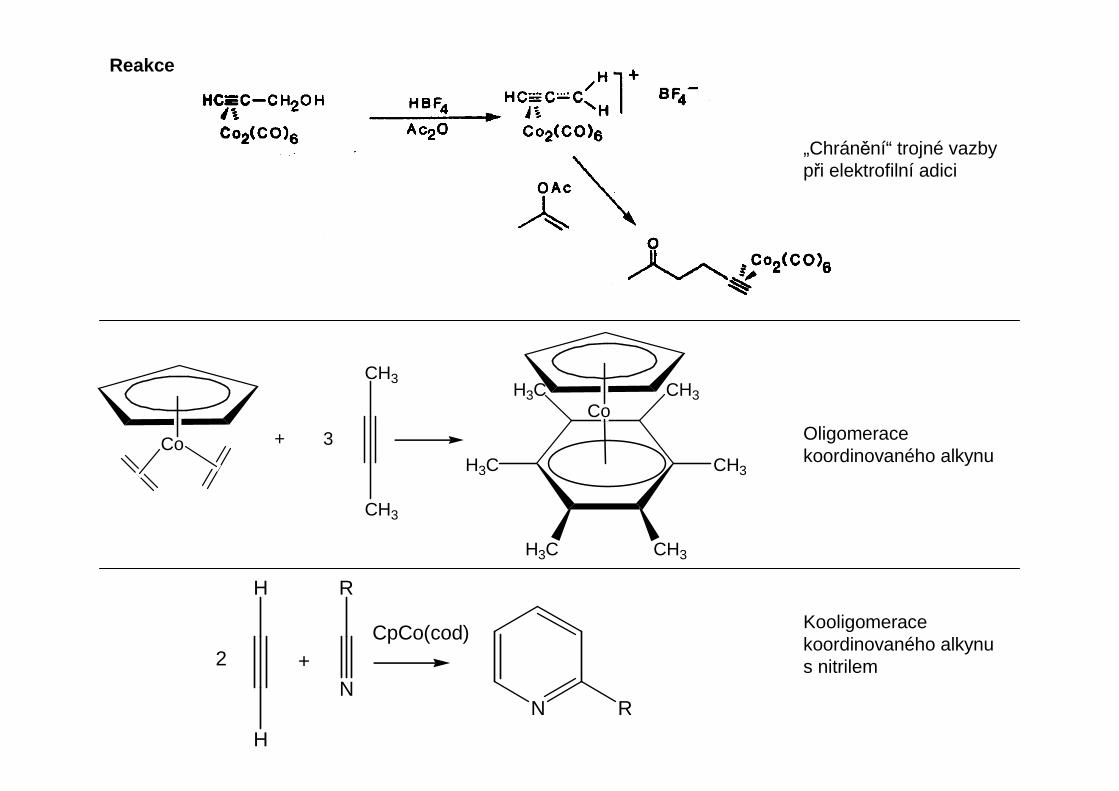
Alkynové komplexy
Jsou podobné olefinovým komplexům, ale vazebné možnosti alkynového ligandu jsou větší.Podobně jako u alkenů můžeme komplexy alkynů někdy považovat za metalacyklopropeny.
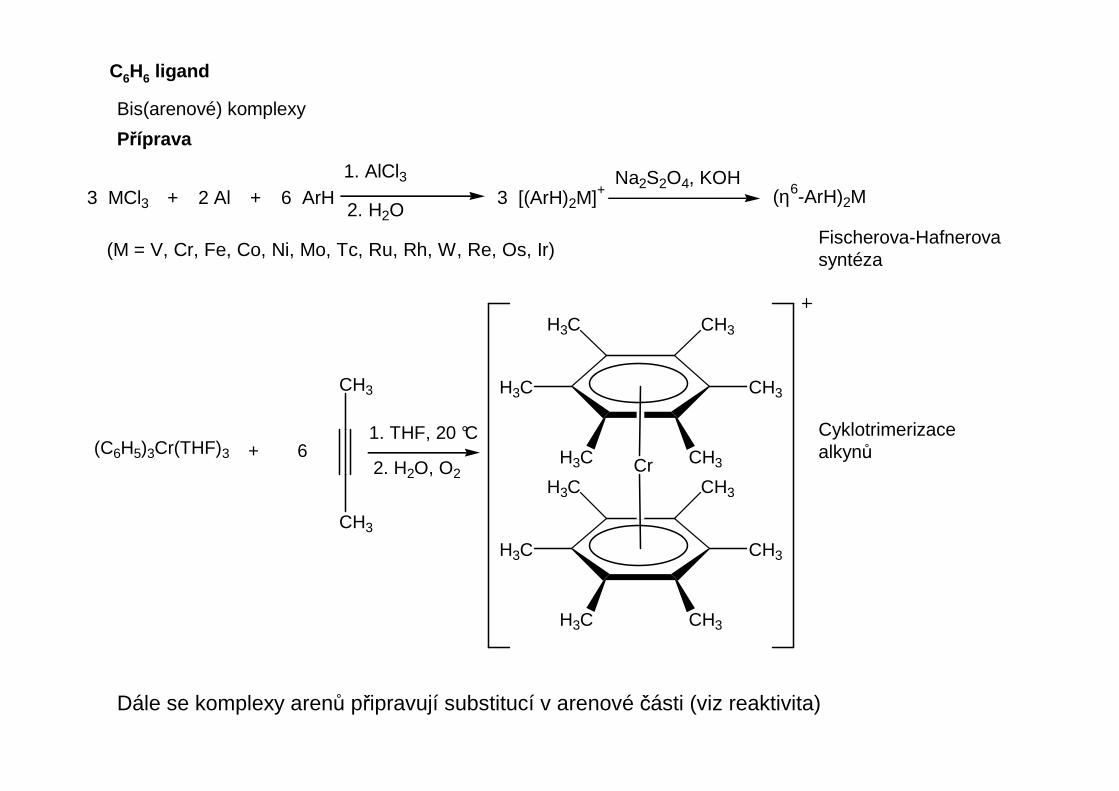
Dále se komplexy arenů připravují substitucí v arenové části (viz reaktivita)
Cr+ 6
CH3
CH3
CH3H3C
H3C CH3
CH3H3C
(C6H5)3Cr(THF)3
CH3H3C
H3C CH3
CH3H3C1. THF, 20 °C
2. H2O, O2
Bis(arenové) komplexy
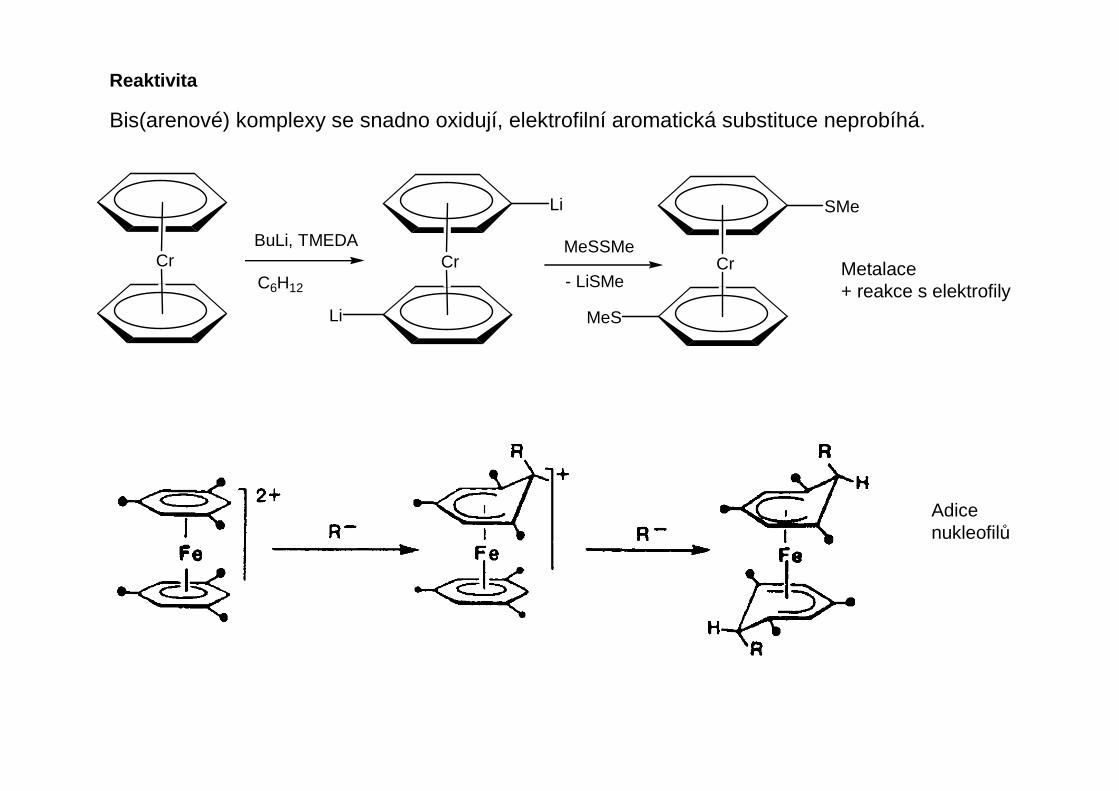
Reaktivita
Bis(arenové) komplexy se snadno oxidují, elektrofilní aromatická substituce neprobíhá.
Cr Cr CrBuLi, TMEDA
C6H12
Li
Li
MeSSMe
- LiSMe
SMe
MeS
Metalace+ reakce s elektrofily
Adicenukleofilů
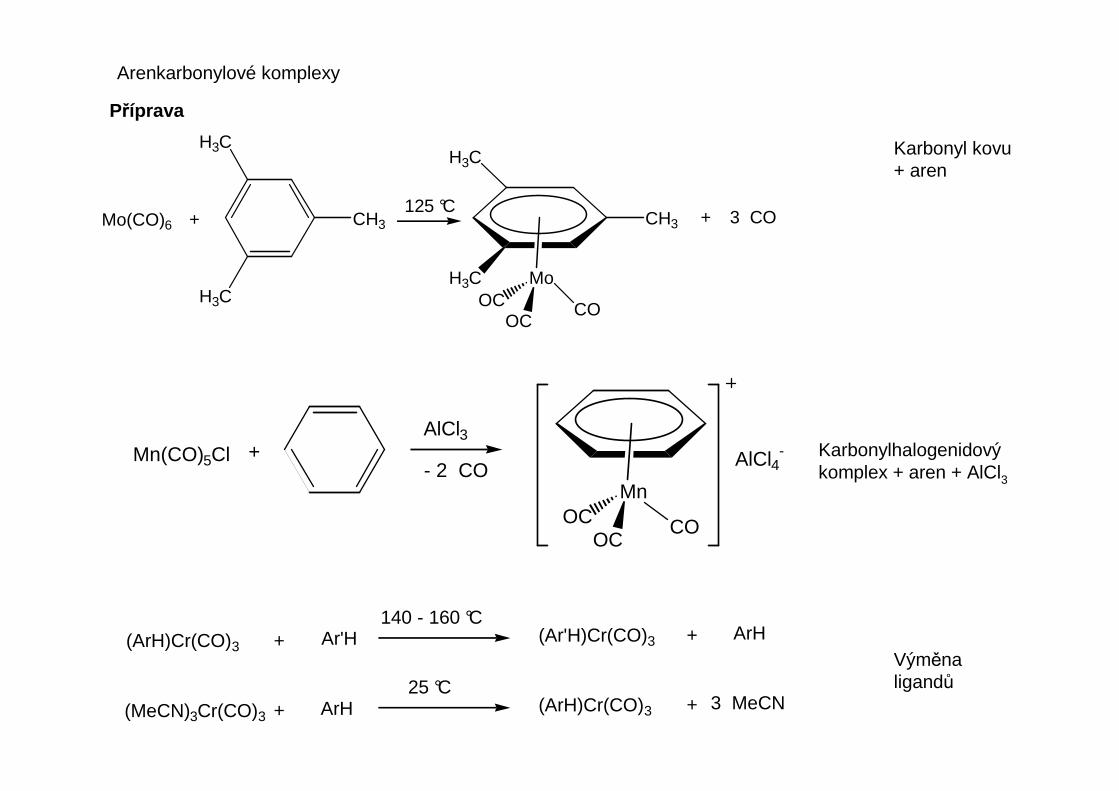
Arenkarbonylové komplexy
Příprava
Karbonyl kovu+ aren
Mo
CH3
H3C
H3C
H3C
H3C
CH3Mo(CO)6 +125 °C
COOC
OC
+ 3 CO
Mn(CO)5Cl +AlCl3
- 2 COMn
COOC
OC
AlCl4-
(ArH)Cr(CO)3 + Ar'H140 - 160 °C
(Ar'H)Cr(CO)3 + ArH
(MeCN)3Cr(CO)3 +25 °C
(ArH)Cr(CO)3 + 3 MeCNArH
Karbonylhalogenidovýkomplex + aren + AlCl3
Výměnaligandů
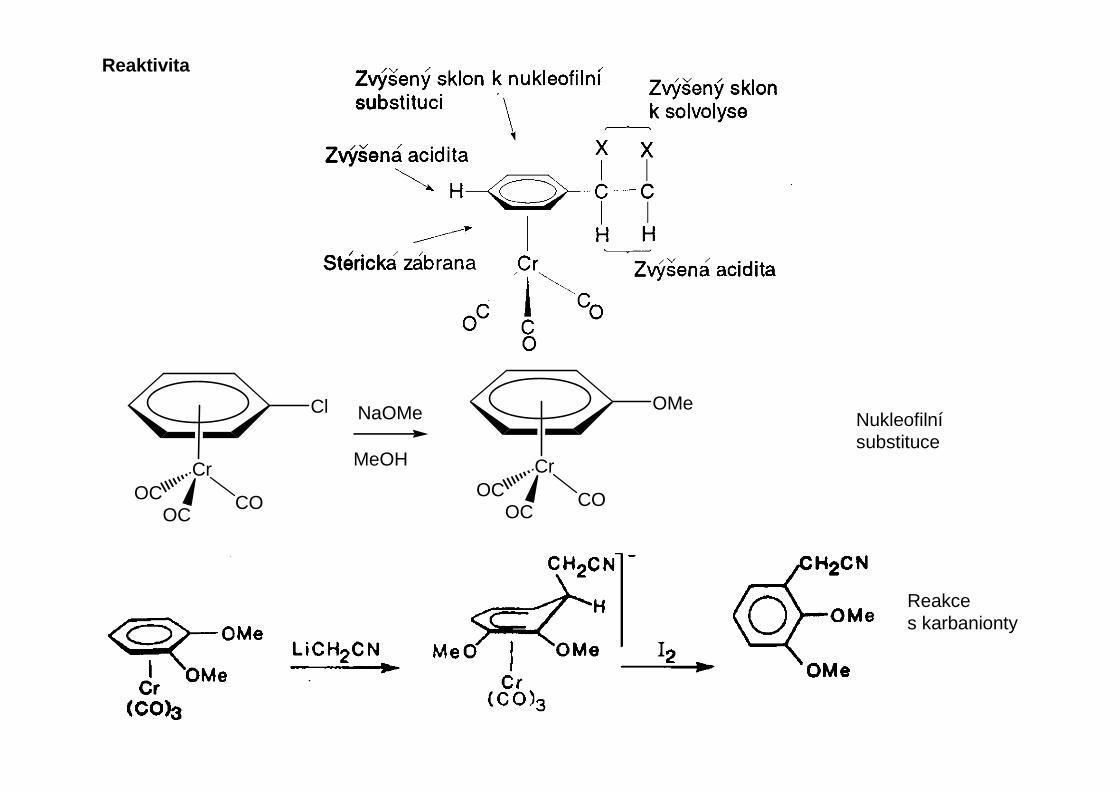
Reaktivita
Cr
COOC
OC
Cl
Cr
COOC
OC
OMeNaOMe
MeOH
Nukleofilnísubstituce
Reakces karbanionty
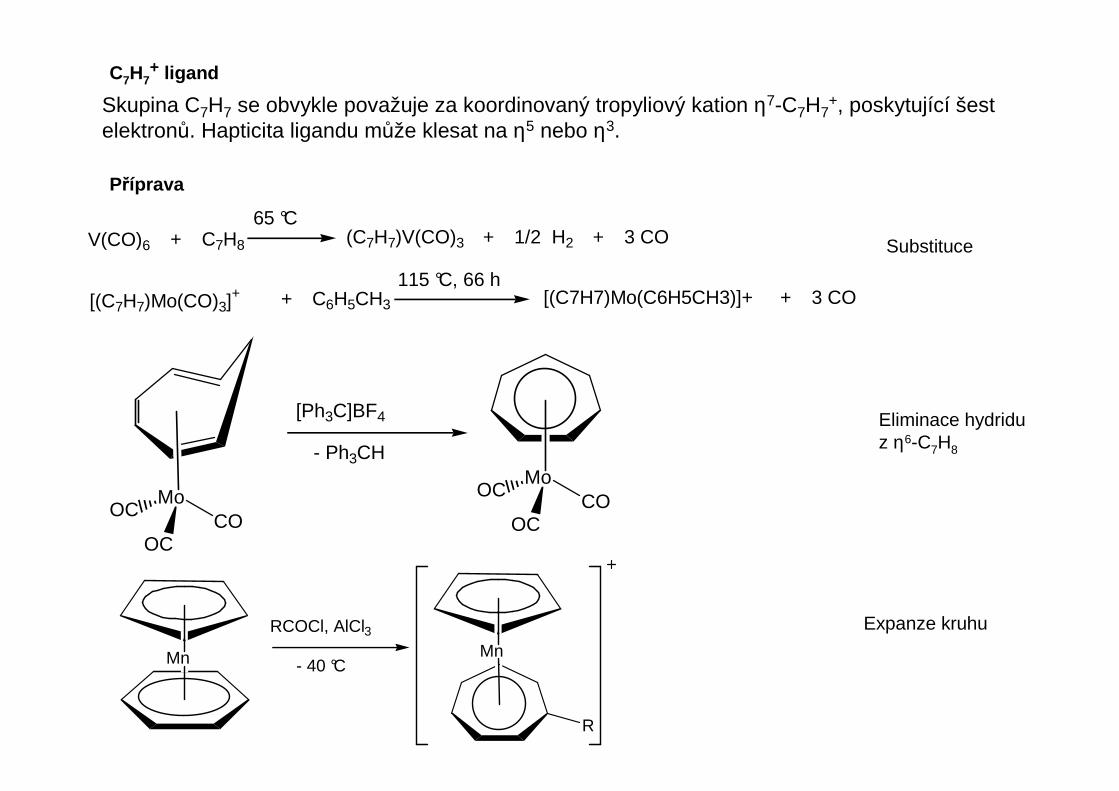
C7H7+ ligand
Příprava
V(CO)6 + C7H8 (C7H7)V(CO)3 + 1/2 H2 + 3 CO65 °C
[(C7H7)Mo(CO)3]+ + C6H5CH3 [(C7H7)Mo(C6H5CH3)]+ + 3 CO115 °C, 66 h
Skupina C7H7 se obvykle považuje za koordinovaný tropyliový kation η7-C7H7+, poskytující šest
elektronů. Hapticita ligandu může klesat na η5 nebo η3.
MoCO
OC
OCMoCO
OC
OC
[Ph3C]BF4
- Ph3CH
Mn Mn
R
RCOCl, AlCl3
- 40 °C
Substituce
Eliminace hydriduz η6-C7H8
Expanze kruhu
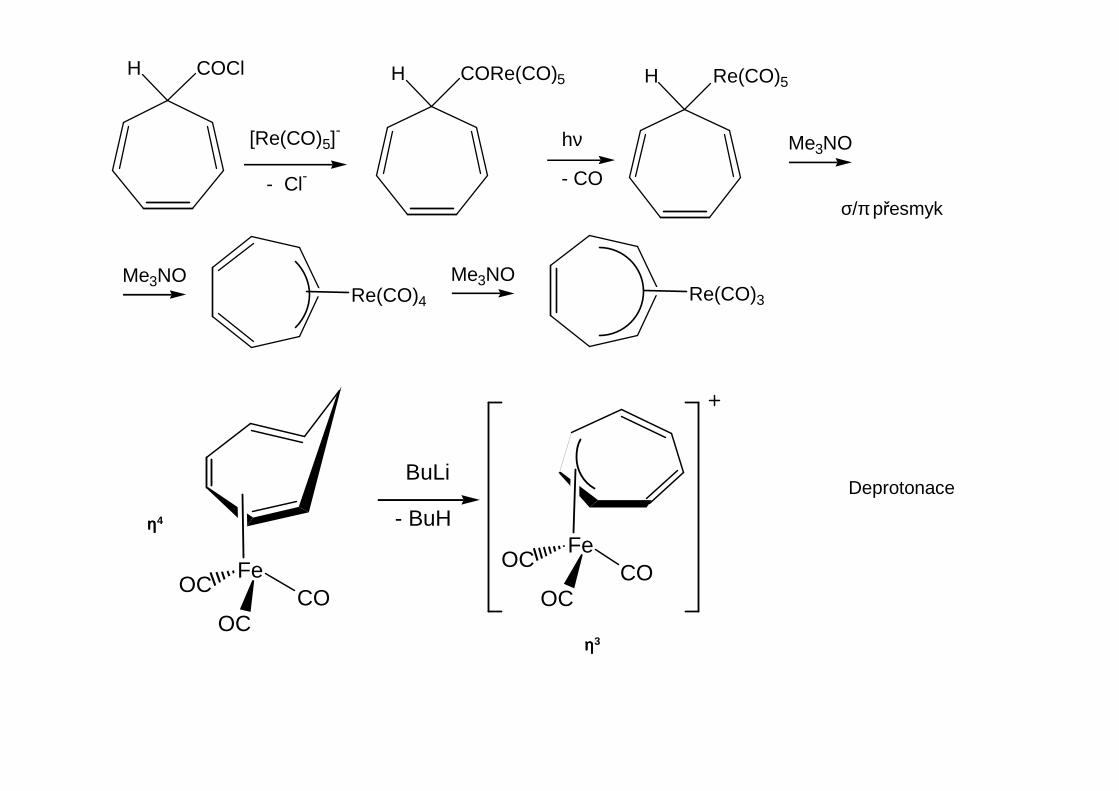
H COCl H CORe(CO)5
[Re(CO)5]-
- Cl-
hν
- CO
H Re(CO)5
Me3NO
Re(CO)4Me3NO Me3NO
Re(CO)3
σ/π přesmyk
Deprotonace
FeCO
OC
OCFeCO
OC
OC
BuLi
- BuHηηηη4
ηηηη3
Reaktivita
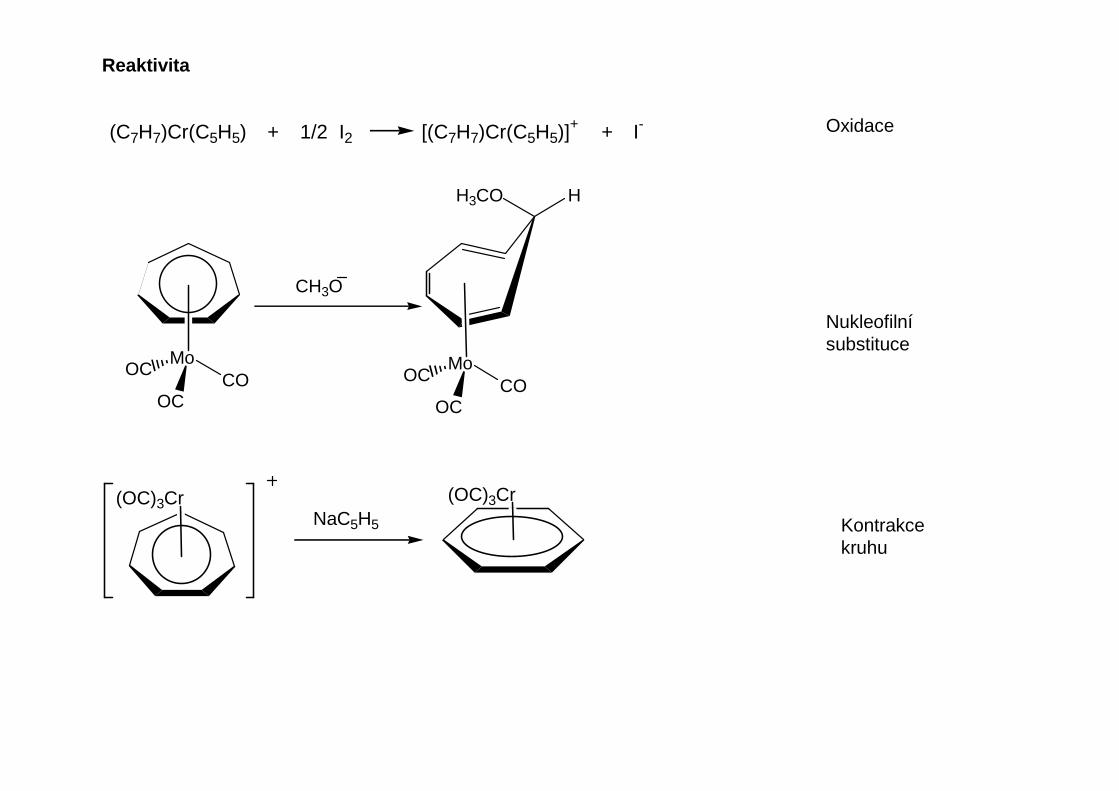
(C7H7)Cr(C5H5) + 1/2 I2 [(C7H7)Cr(C5H5)]+ + I-
MoCO
OC
OC MoCO
OC
OC
CH3O
H3CO H
(OC)3Cr(OC)3CrNaC5H5
Nukleofilnísubstituce
Oxidace
Kontrakcekruhu
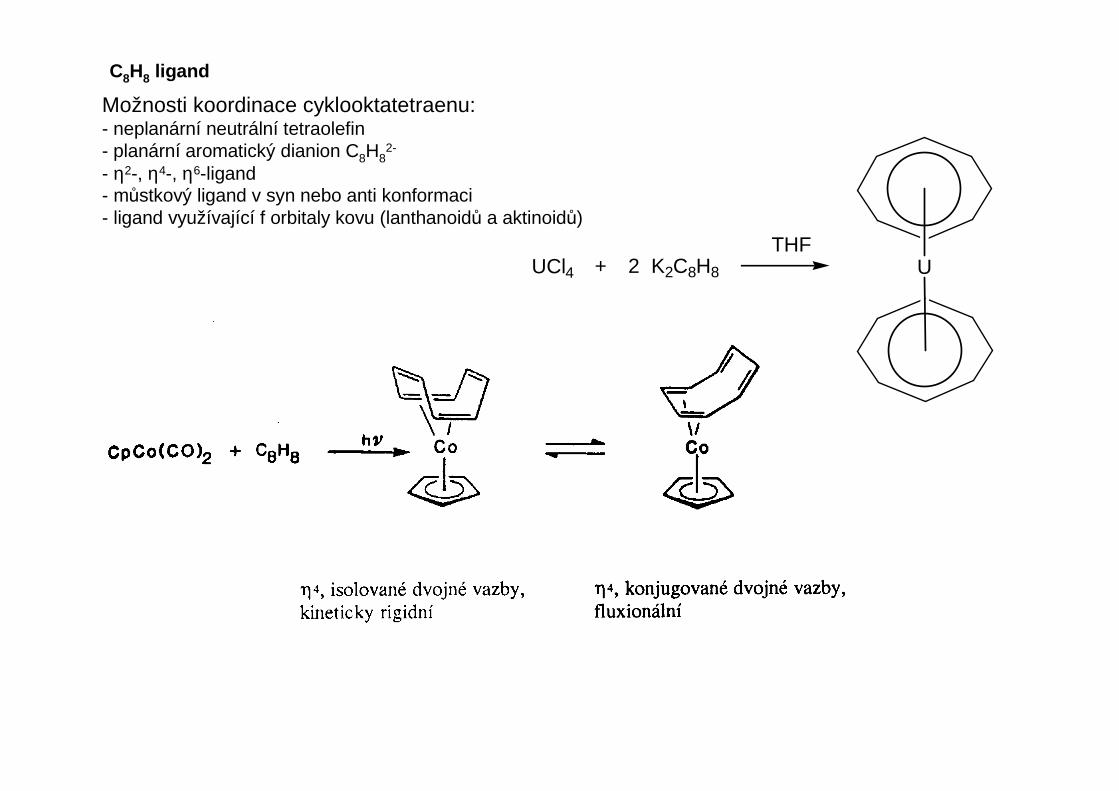
C8H8 ligand
Možnosti koordinace cyklooktatetraenu:- neplanární neutrální tetraolefin- planární aromatický dianion C8H8
2-
- η2-, η4-, η6-ligand- můstkový ligand v syn nebo anti konformaci- ligand využívající f orbitaly kovu (lanthanoidů a aktinoidů)
UUCl4 + 2 K2C8H8
THF
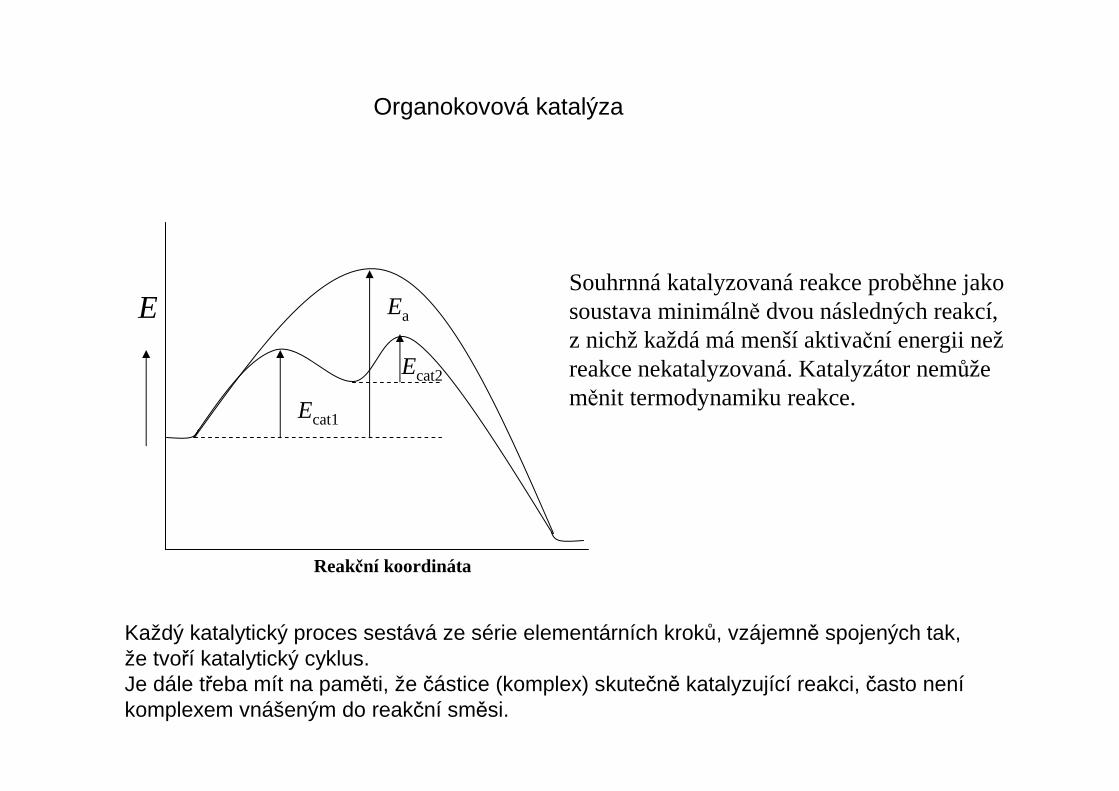
Organokovová katalýza
Souhrnná katalyzovaná reakce proběhne jakosoustava minimálně dvou následných reakcí,z nichž každá má menší aktivační energii nežreakce nekatalyzovaná. Katalyzátor nemůžeměnit termodynamiku reakce.
E Ea
Ecat1
Reakční koordináta
Ecat2
Každý katalytický proces sestává ze série elementárních kroků, vzájemně spojených tak,že tvoří katalytický cyklus.Je dále třeba mít na paměti, že částice (komplex) skutečně katalyzující reakci, často neníkomplexem vnášeným do reakční směsi.
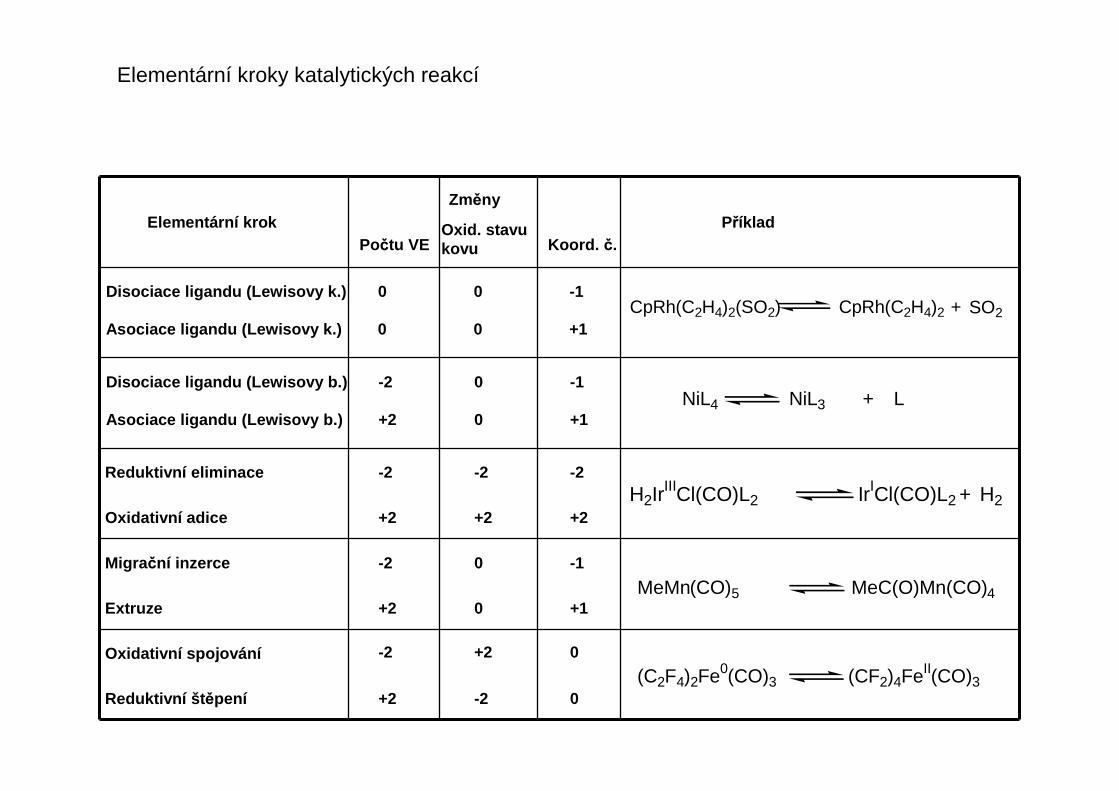
Jak fungují komplexy přechodných kovů v katalýze ?
Koordinace substrátu(ů) k přechodnému kovu přivádí reakční partnery do těsné blízkosti
Koordinace k přechodnému kovu mění distribuci elektronů v ligandu a tím jej aktivuje pro reakci
Koordinací organického substrátu se usnadňuje jeho atak nukleofilem
Rozlišujeme (dnes spíše z historických důvodů)
Heterogenní katalýzu: katalyticky aktivní koordinační místa jsou na fázovém rozhraní s/l nebo s/g,t.j. aktivní jsou jen povrchové atomy pevného katalyzátoru
Homogenní katalýzu: Katalyticky aktivní komplexy kovů jsou rozpustné v reakčním prostředí,katalýza probíhá v jediné fázi - kapalné
Platí pro diamagnetické částice v koncentracích dokazatelných spektroskopicky neboanalýzou kinetických dat.
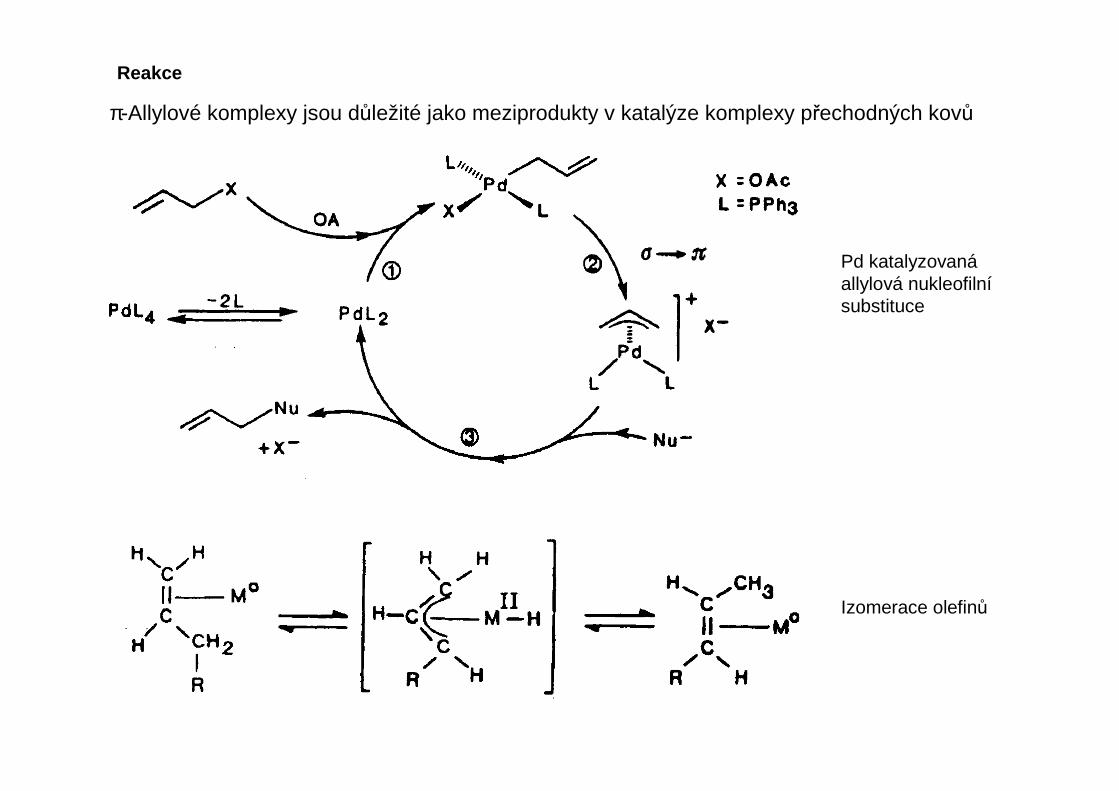
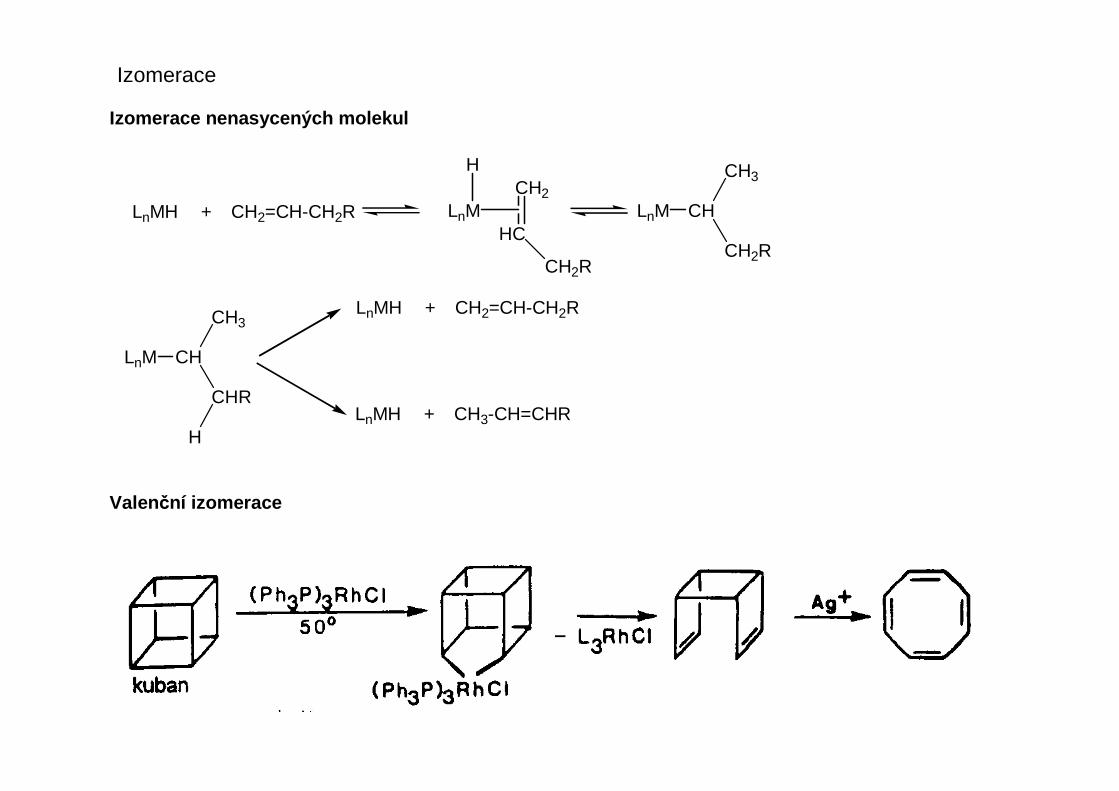
Izomerace
Izomerace nenasycených molekul
LnMH + CH2=CH-CH2RCH2
HC
CH2R
LnM
H
LnM CH
CH3
CH2R
LnM CH
CH3
CHR
H
LnMH + CH2=CH-CH2R
LnMH + CH3-CH=CHR
Valenční izomerace
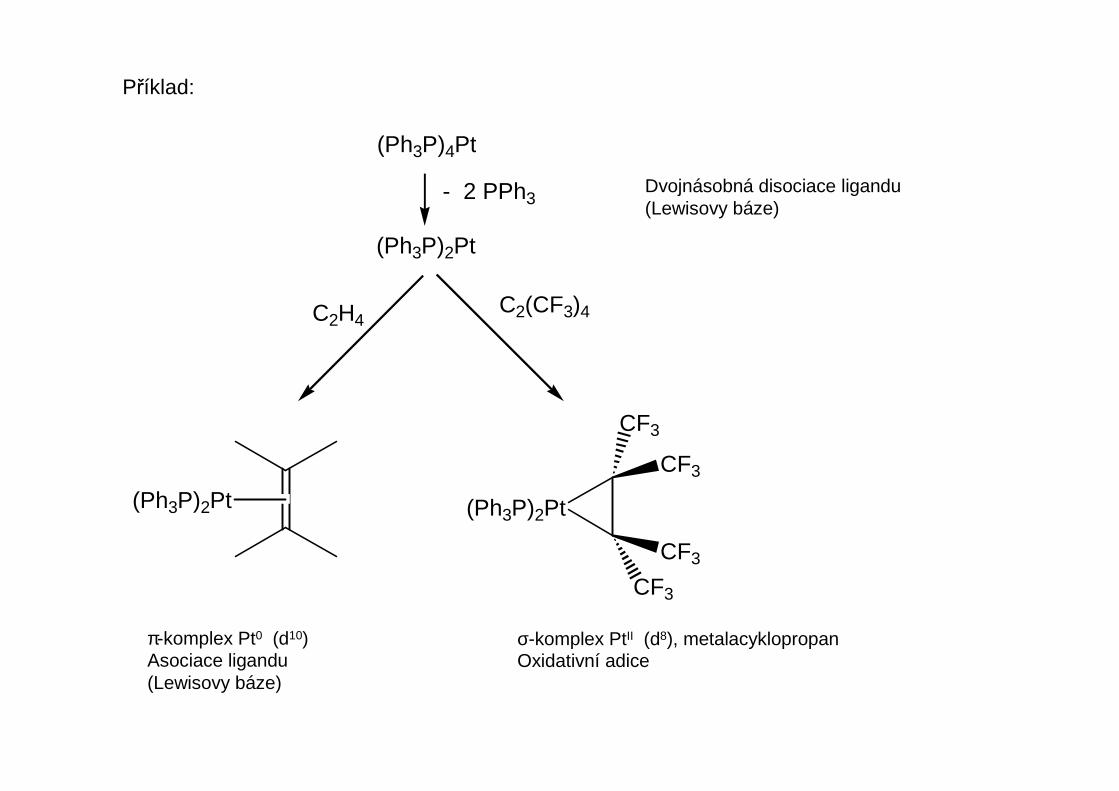
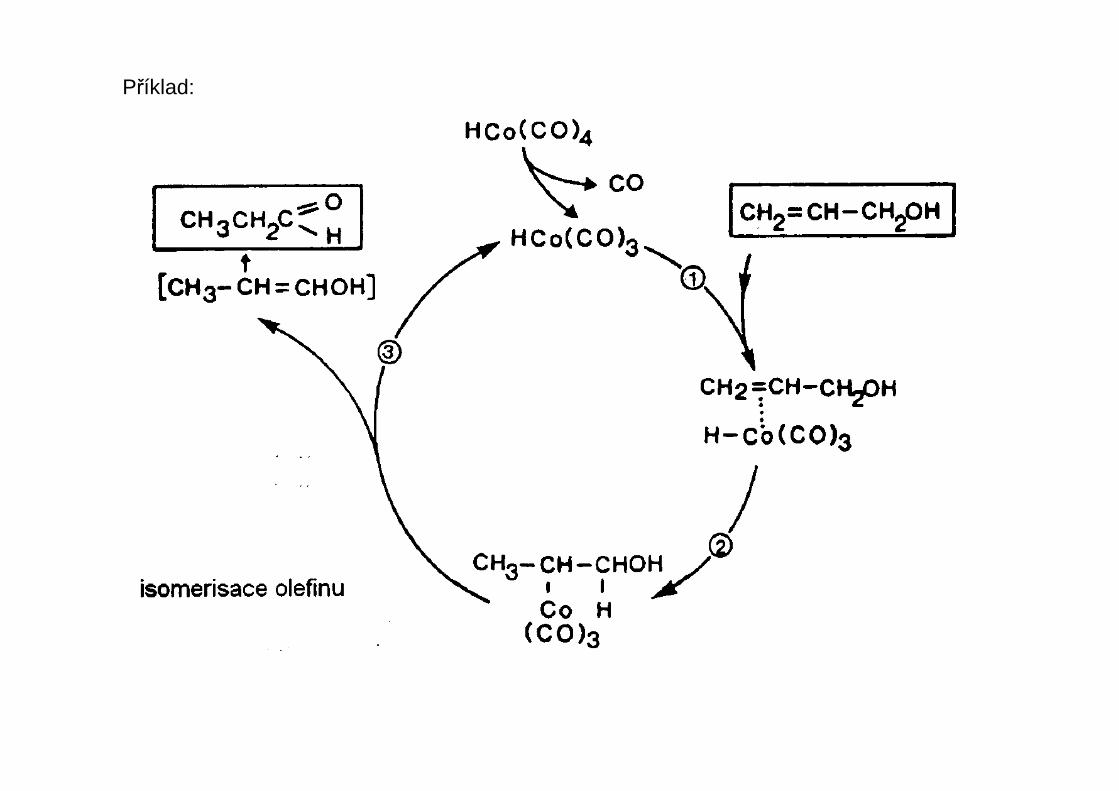
Příklad:
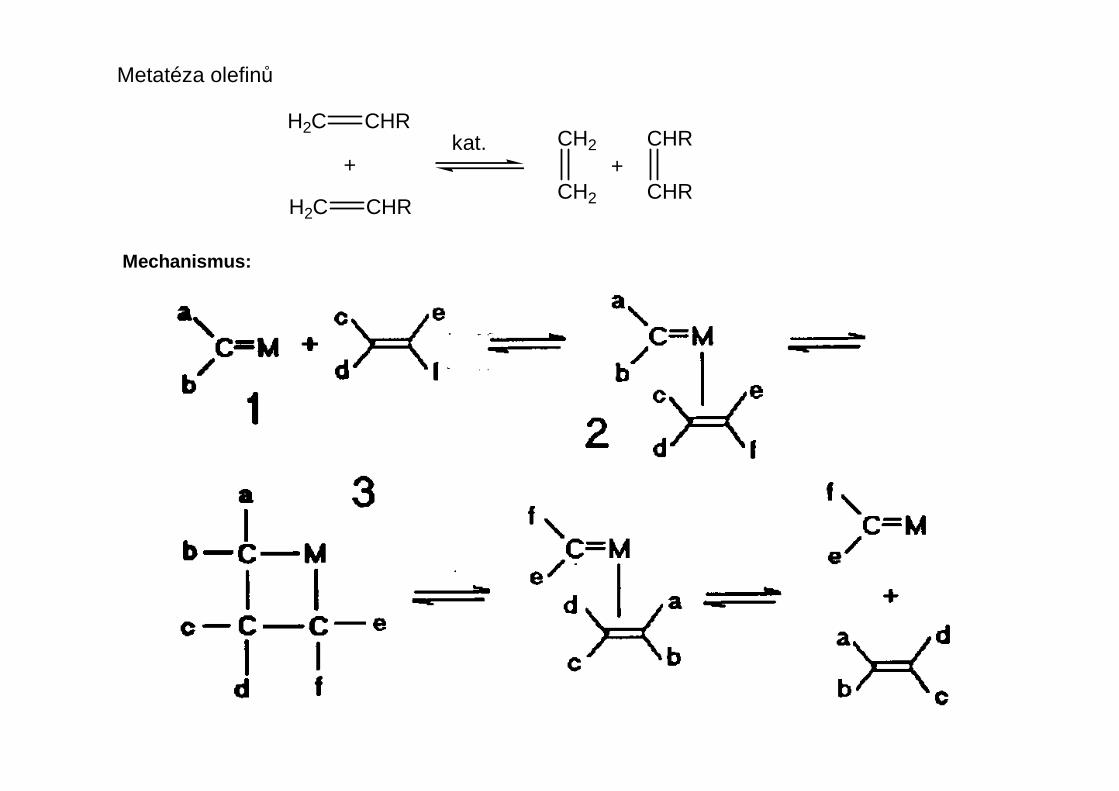
Metatéza olefinů
H2C CHR
H2C CHR
+kat. CH2
CH2
CHR
CHR+
Mechanismus:
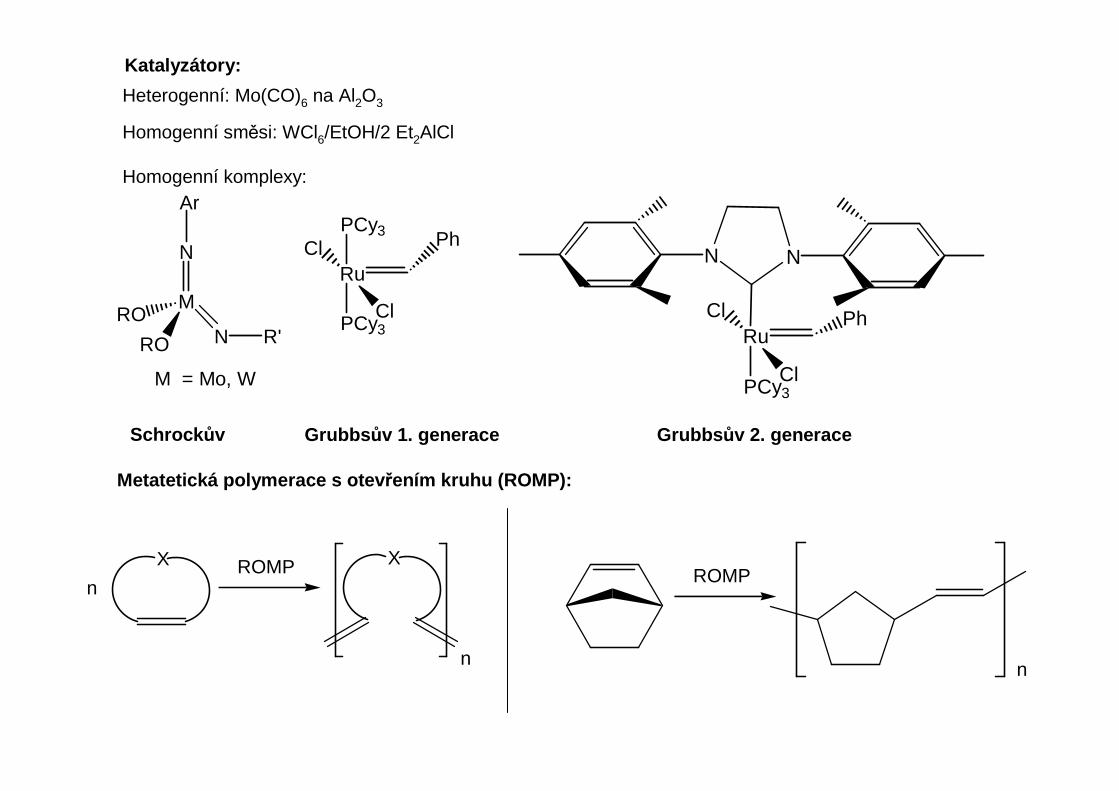
Katalyzátory:
Heterogenní: Mo(CO)6 na Al2O3
Homogenní směsi: WCl6/EtOH/2 Et2AlCl
Homogenní komplexy:
M
N
NRO
RO
Ar
R'
M = Mo, W
Ru
PCy3
PCy3Cl
Cl Ph
Ru
PCy3Cl
Cl Ph
NN
Schrock ův Grubbs ův 1. generace Grubbs ův 2. generace
Metatetická polymerace s otev řením kruhu (ROMP):
X ROMP X
n
n
n
ROMP
Oligomerace a polymerace
Trocha historie
V padesátých letech minulého století se K. Ziegler zabýval možnostmi, jak zvýšit molekulovouhmotnost při polymeraci olefinů s použitím trialkylhliníků. Reakci konkuruje dehydroaluminacea tvoří se oligomery až polymery s maximální délkou řetězce kolem C200.
V jednom pokusu byl překvapivě získán výlučně dimer ethylenu. Zjistilo se, že příčinou byla stopanikelnatých solí v autoklávu po jeho vymývání koncentrovanou HCl („nickel effect“). Rozsáhléstudium kat. systémů sůl přechodného kovu/R3Al pak vedlo (1955) k objevu polymerace olefinůtzv. Zieglerovým-Nattovým procesem. Vzniklý nízkotlaký polyethylen je přísně lineární.
R2AlC2H5 + (n-1) CH2=CH2
90 - 120 °C, 100 barR2Al(CH2CH2)nH
R2Al(CH2CH2)nR' R2AlH + CH2=CHR'
CH2=CH2 polyethylenTiCl4/Et3Al, 25 °C, 1 bar
Reakce však neprobíhá zcela v homogenním prostředí, katalytickou částicí je pravděpodobněalkylovaný vláknitý TiCl3
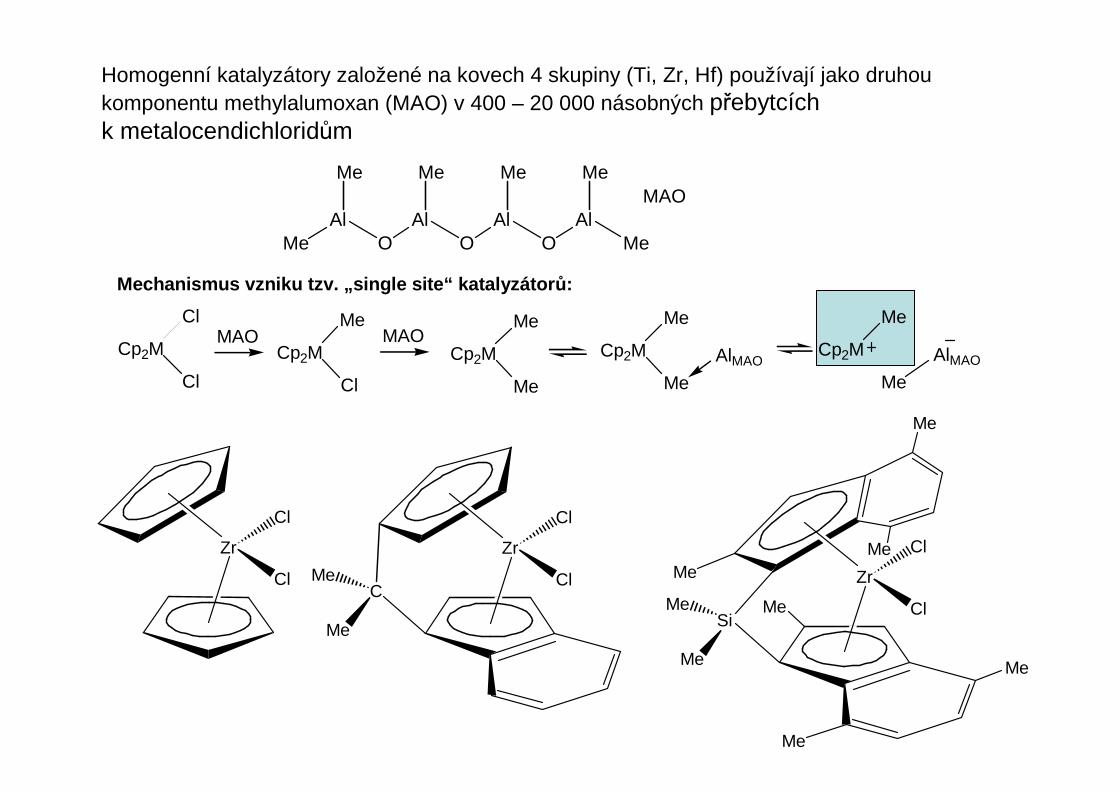
Homogenní katalyzátory založené na kovech 4 skupiny (Ti, Zr, Hf) používají jako druhoukomponentu methylalumoxan (MAO) v 400 – 20 000 násobných přebytcích k metalocendichloridům
Mechanismus vzniku tzv. „single site“ katalyzátor ů:
MeAl
OAl
OAl
OAl
Me
Me Me Me MeMAO
Cp2M
Cl
Cl
Cp2M
Me
Cl
MAOCp2M
Me
Me
MAOCp2M
Me
Me
AlMAOCp2M
Me
Me
AlMAO
Zr
Cl
Cl
Zr
Cl
Cl
C
Me
Me Zr
Cl
Cl
Si
Me
Me
Me
Me
Me
Me
Me
Me
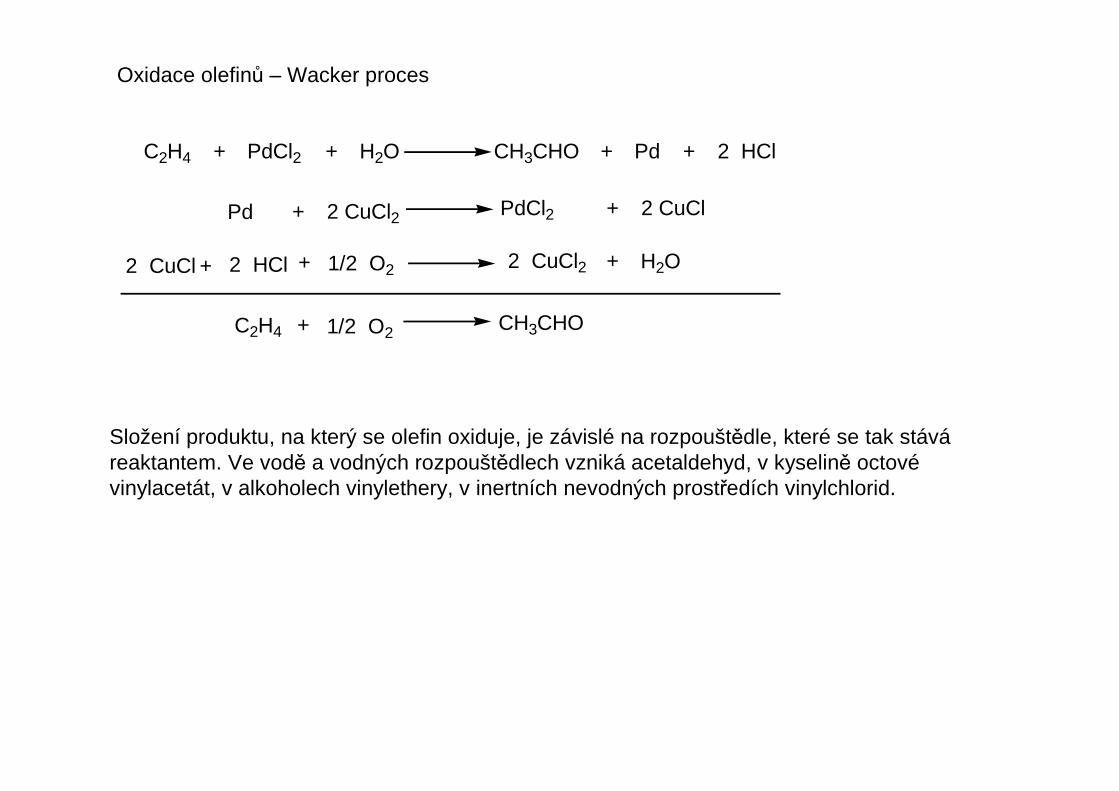
Oxidace olefinů – Wacker proces
C2H4 + + H2O CH3CHO + Pd + 2 HClPdCl2
Pd + 2 CuCl2 PdCl2 + 2 CuCl
2 CuCl + 2 HCl + 1/2 O2 2 CuCl2 + H2O
C2H4 + 1/2 O2 CH3CHO
Složení produktu, na který se olefin oxiduje, je závislé na rozpouštědle, které se tak stáváreaktantem. Ve vodě a vodných rozpouštědlech vzniká acetaldehyd, v kyselině octovévinylacetát, v alkoholech vinylethery, v inertních nevodných prostředích vinylchlorid.
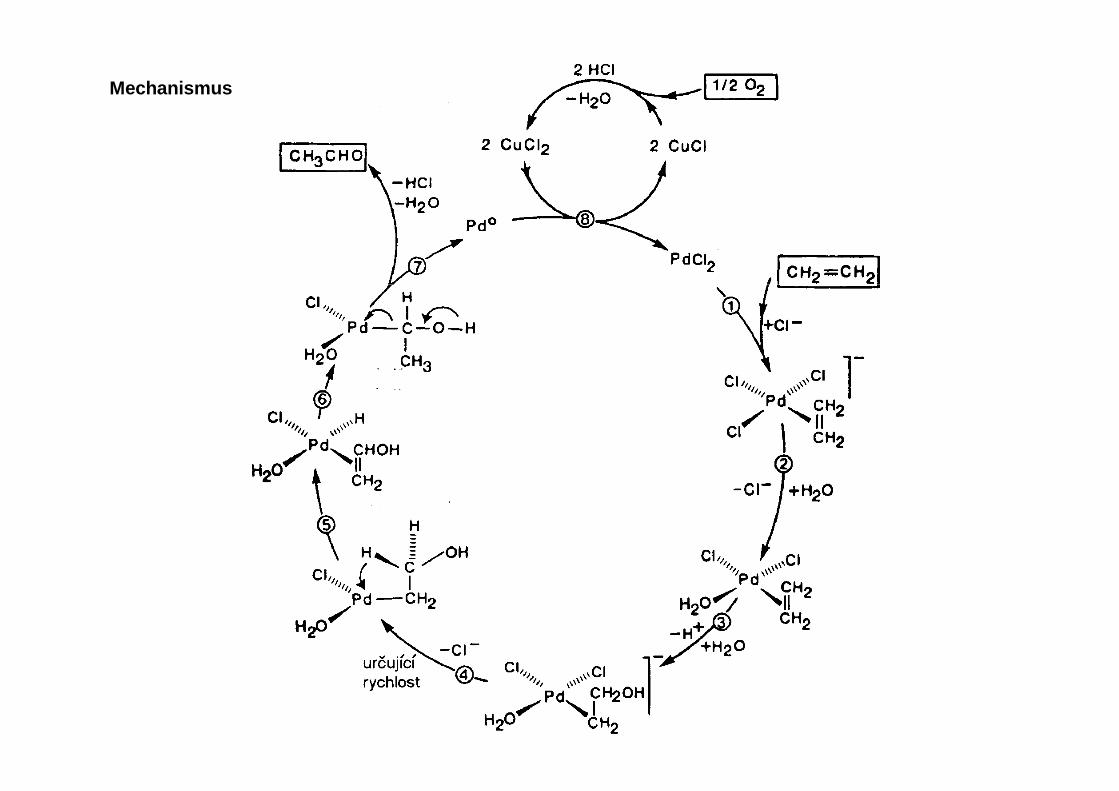
Mechanismus
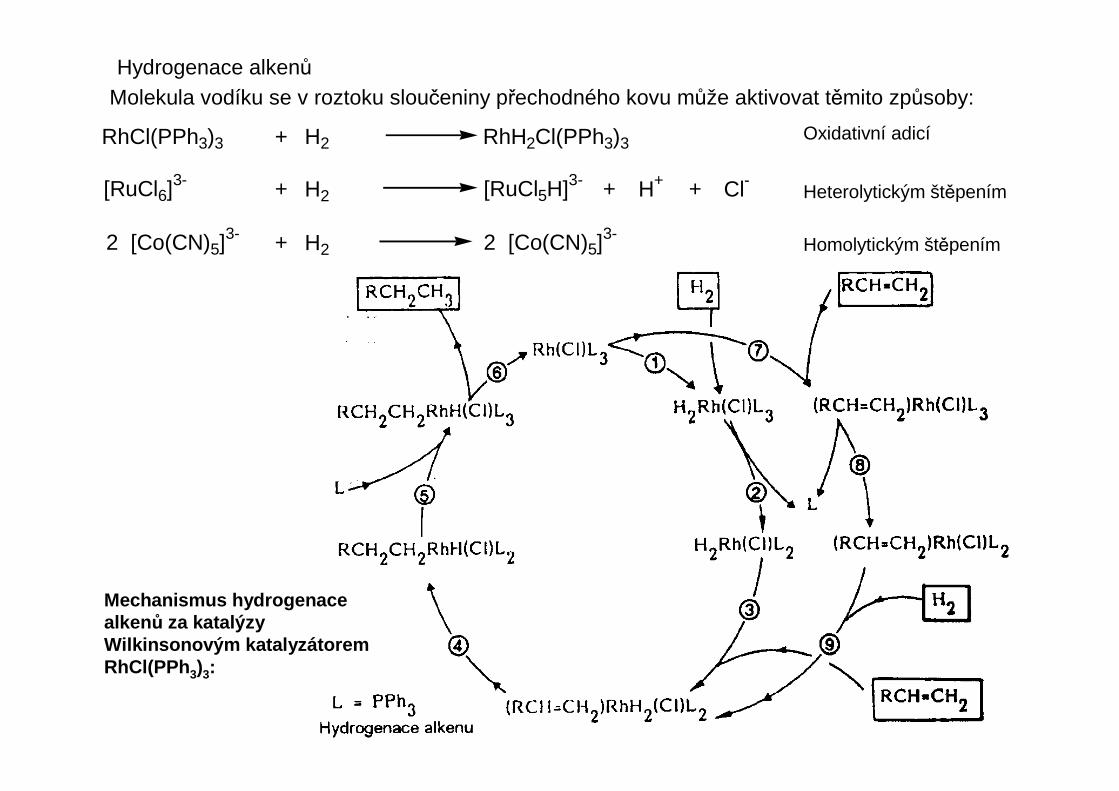
Hydrogenace alkenůMolekula vodíku se v roztoku sloučeniny přechodného kovu může aktivovat těmito způsoby:
RhCl(PPh3)3 + H2 RhH2Cl(PPh3)3
[RuCl6]3- + H2 [RuCl5H]3- + H+ + Cl-
2 [Co(CN)5]3- + H2 2 [Co(CN)5]3-
Oxidativní adicí
Heterolytickým štěpením
Homolytickým štěpením
Mechanismus hydrogenacealkenů za katalýzyWilkinsonovým katalyzátoremRhCl(PPh 3)3:
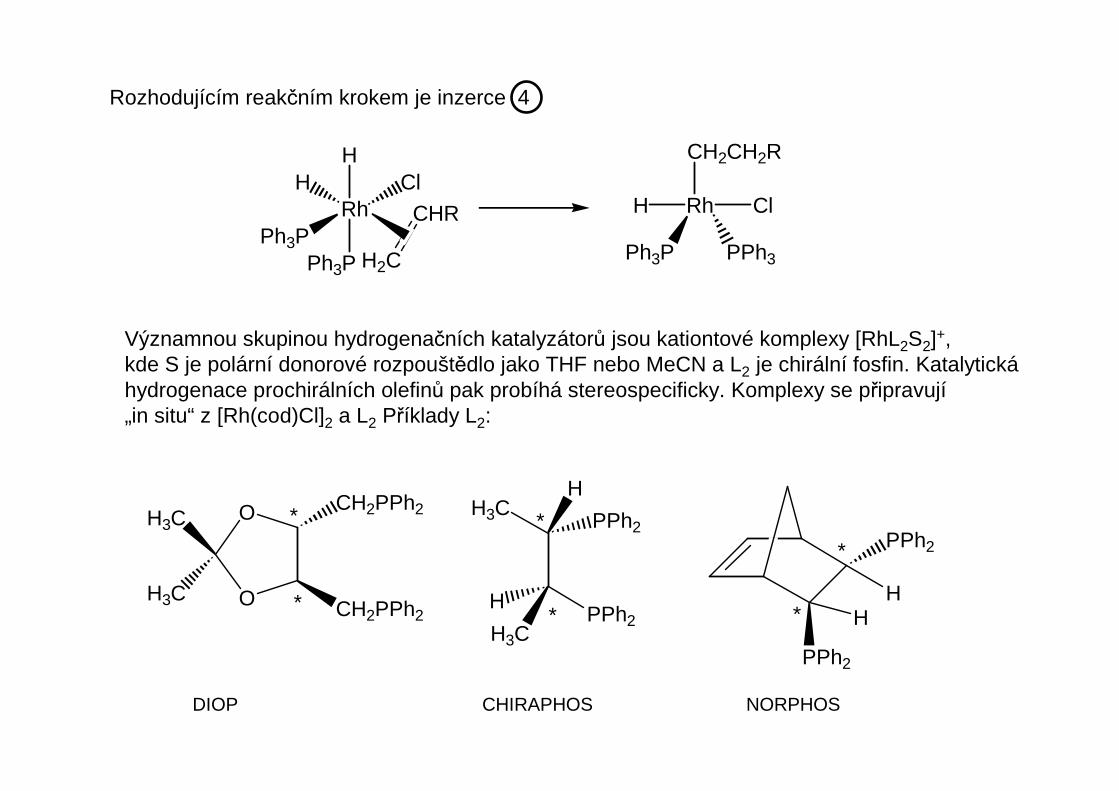
Rozhodujícím reakčním krokem je inzerce 4
Rh
H
Ph3P
H Cl
Ph3PH2C
CHR RhH
CH2CH2R
Cl
Ph3P PPh3
Významnou skupinou hydrogenačních katalyzátorů jsou kationtové komplexy [RhL2S2]+,kde S je polární donorové rozpouštědlo jako THF nebo MeCN a L2 je chirální fosfin. Katalytickáhydrogenace prochirálních olefinů pak probíhá stereospecificky. Komplexy se připravují„in situ“ z [Rh(cod)Cl]2 a L2 Příklady L2:
O
O
H3C
H3C
CH2PPh2
CH2PPh2
*
*
H3C
PPh2
H
H3C
PPh2
H
*
*H
H
PPh2
PPh2*
*
DIOP CHIRAPHOS NORPHOS
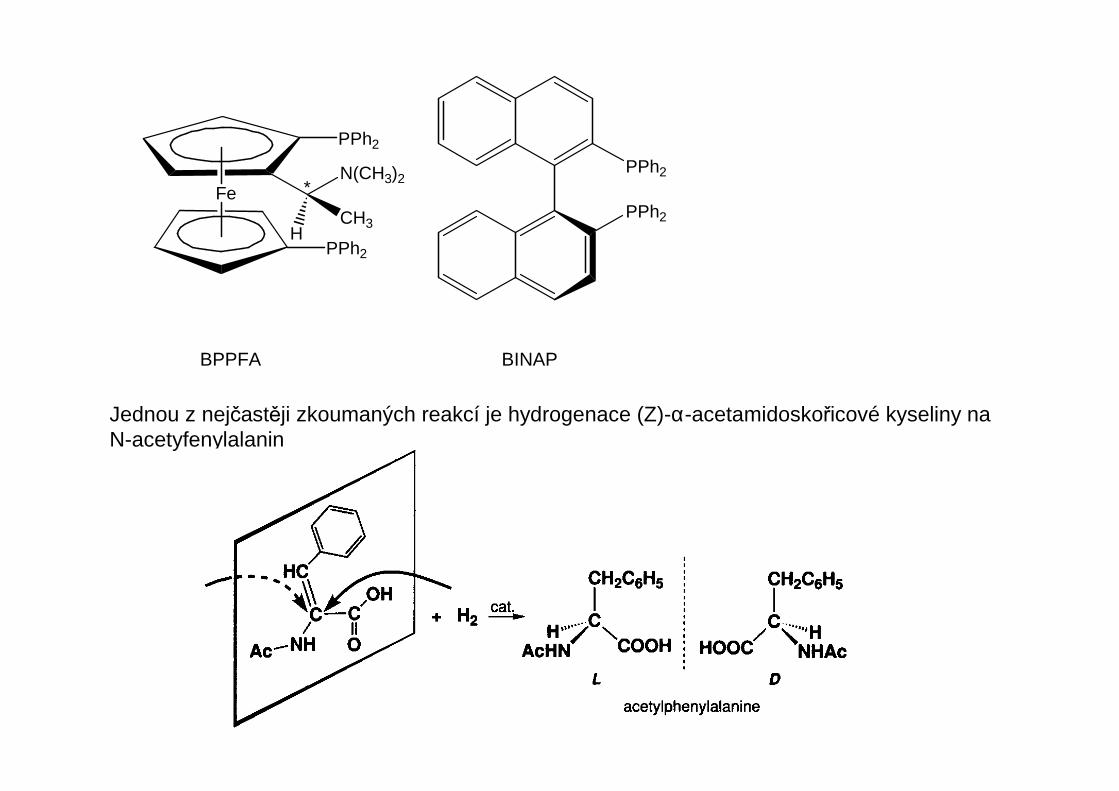
Fe
PPh2
PPh2
N(CH3)2
HCH3
*PPh2
PPh2
BPPFA BINAP
Jednou z nejčastěji zkoumaných reakcí je hydrogenace (Z)-α-acetamidoskořicové kyseliny naN-acetyfenylalanin
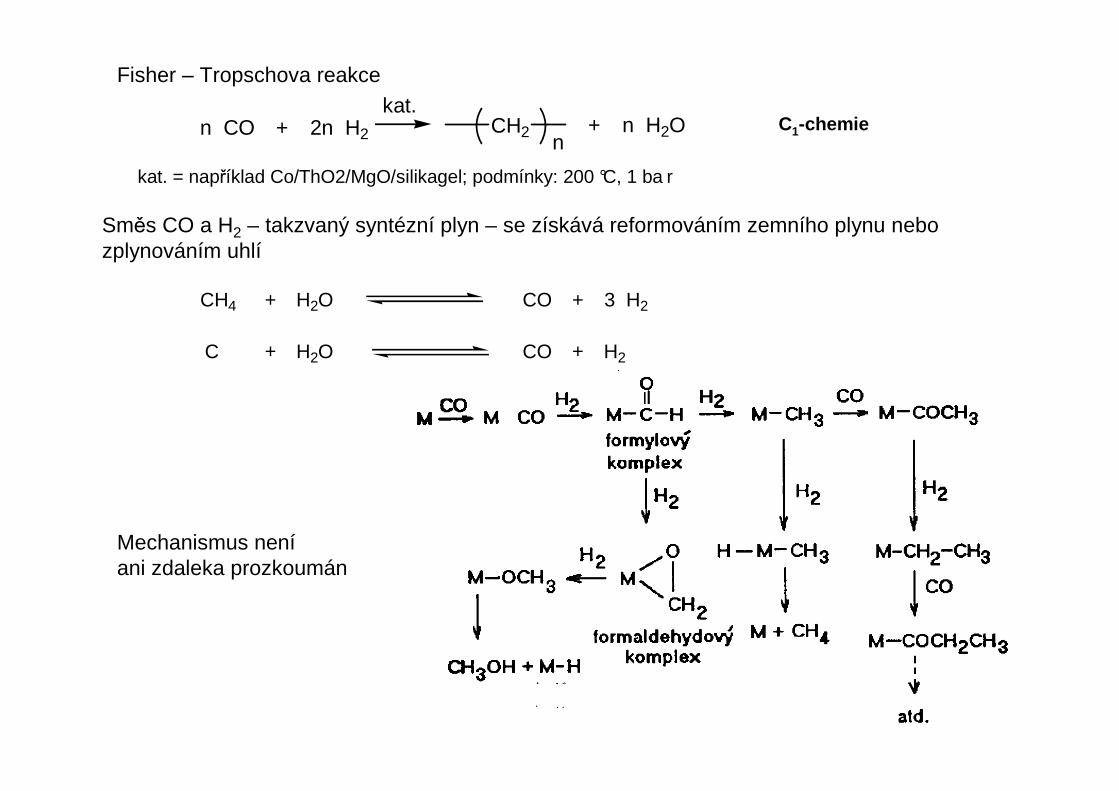
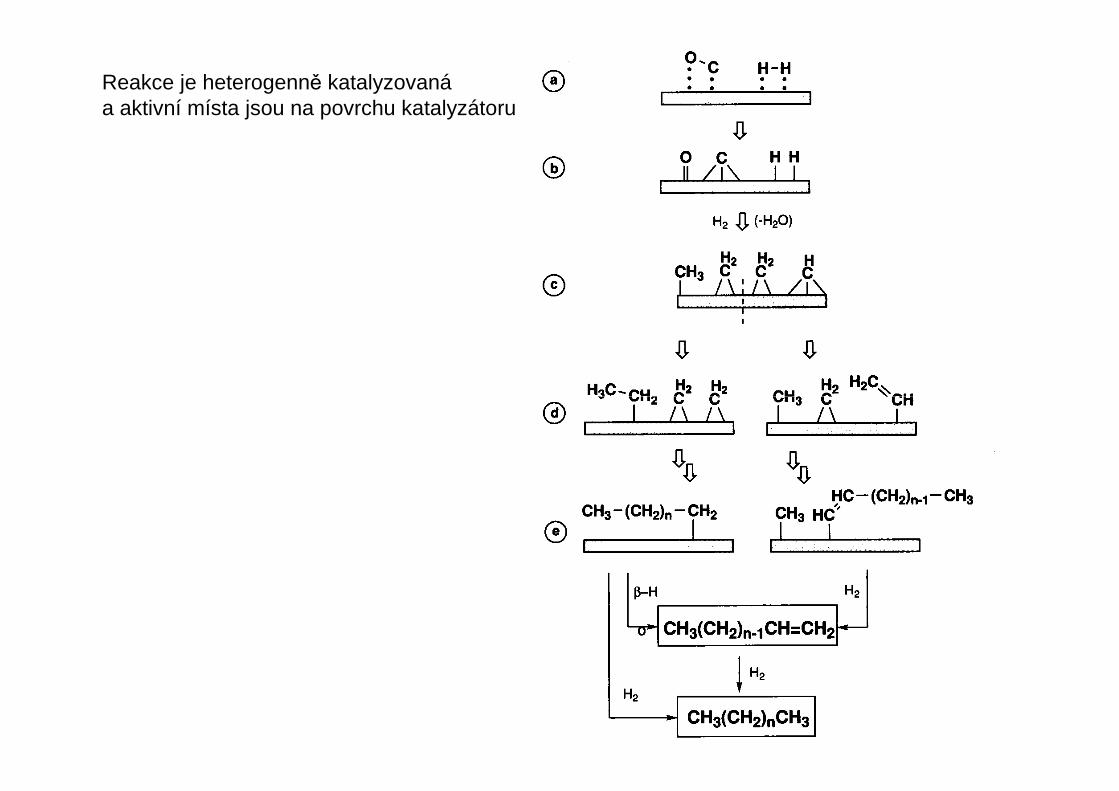
Fisher – Tropschova reakce
n CO + 2n H2 + n H2OCH2n
kat.C1-chemie
Směs CO a H2 – takzvaný syntézní plyn – se získává reformováním zemního plynu nebozplynováním uhlí
CH4 + H2O CO + 3 H2
C + H2O CO + H2
Mechanismus neníani zdaleka prozkoumán
kat. = například Co/ThO2/MgO/silikagel; podmínky: 200 °C, 1 ba r
Reakce je heterogenně katalyzovanáa aktivní místa jsou na povrchu katalyzátoru
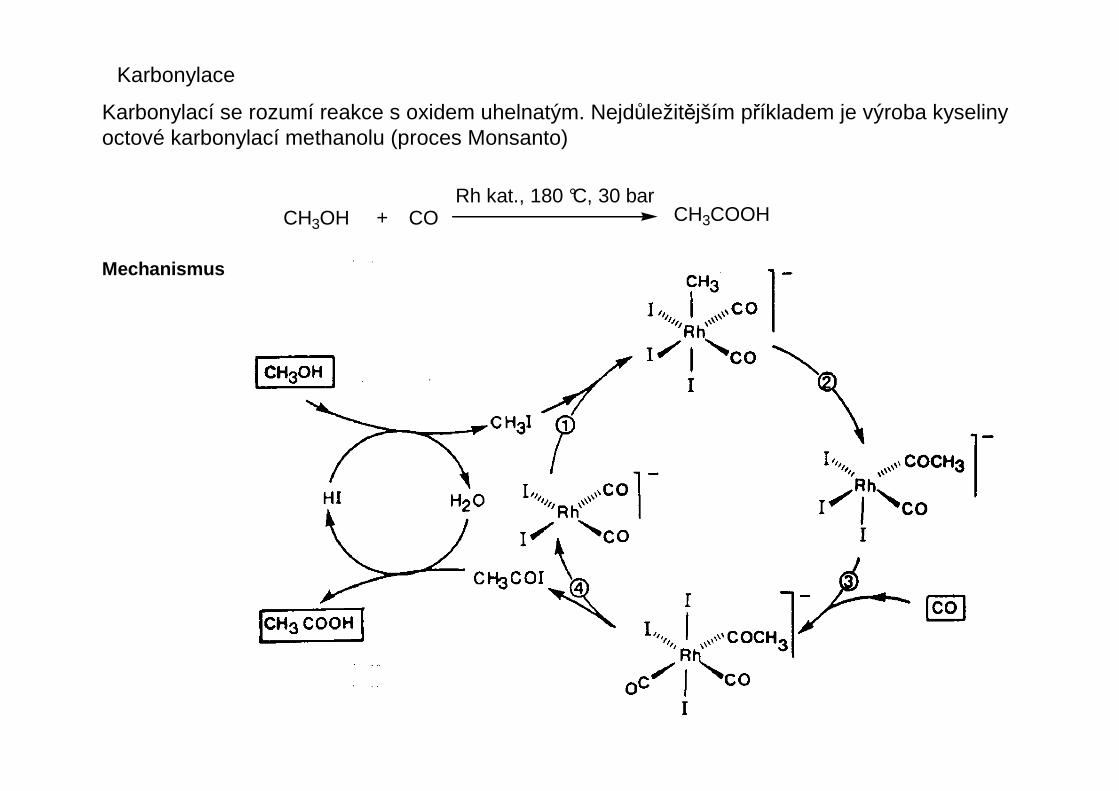
Karbonylace
Karbonylací se rozumí reakce s oxidem uhelnatým. Nejdůležitějším příkladem je výroba kyselinyoctové karbonylací methanolu (proces Monsanto)
CH3OH + CORh kat., 180 °C, 30 bar
CH3COOH
Mechanismus
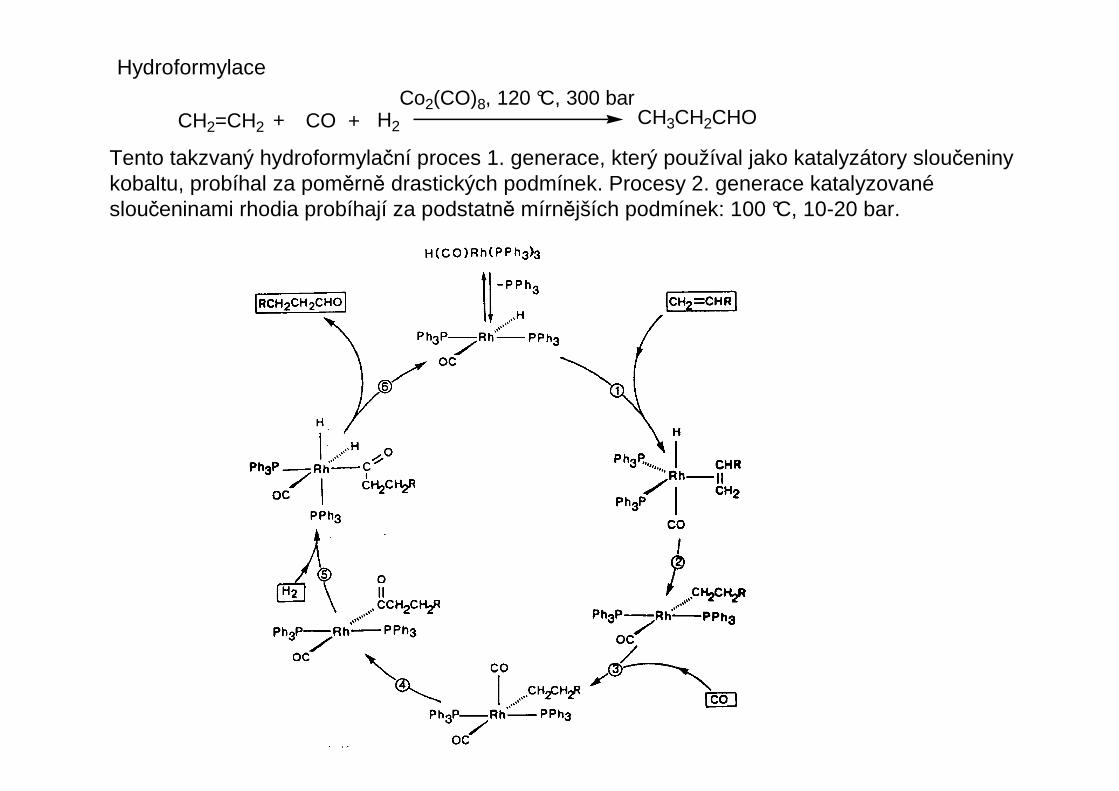
Hydroformylace
CH2=CH2 + COCo2(CO)8, 120 °C, 300 bar
CH3CH2CHO+ H2
Tento takzvaný hydroformylační proces 1. generace, který používal jako katalyzátory sloučeninykobaltu, probíhal za poměrně drastických podmínek. Procesy 2. generace katalyzovanésloučeninami rhodia probíhají za podstatně mírnějších podmínek: 100 °C, 10-20 bar.
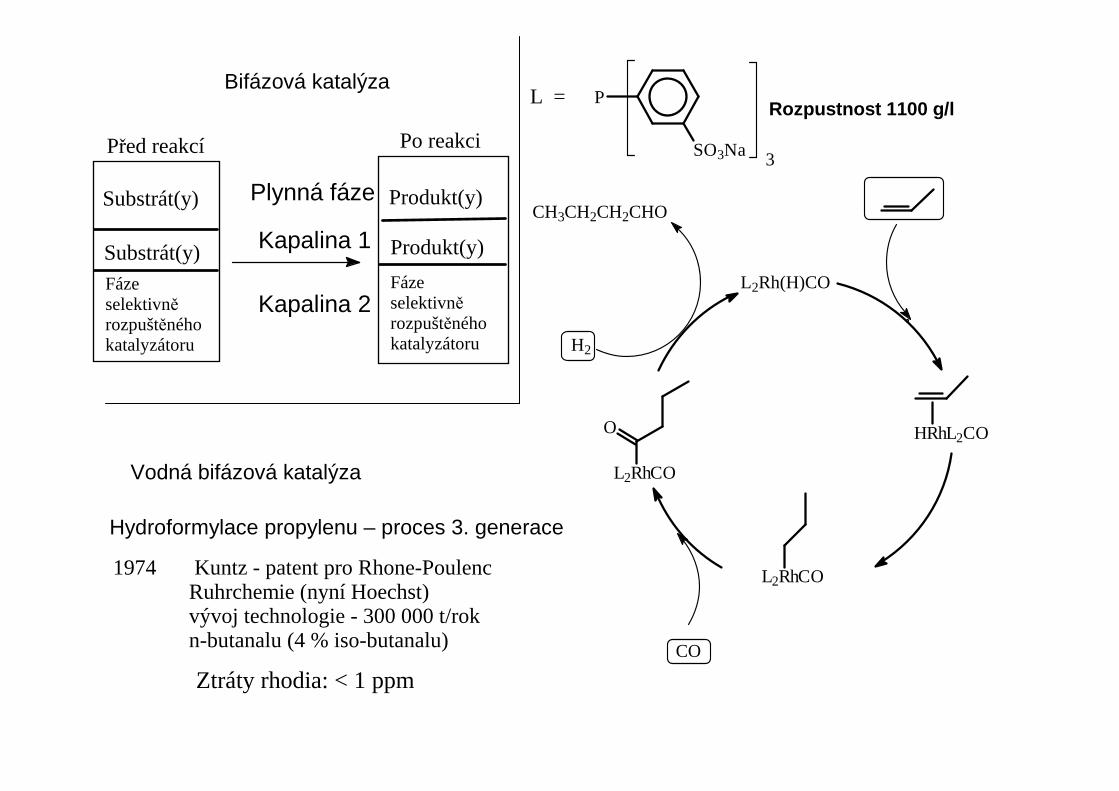
3SO3Na
PL =
H2
L2Rh(H)CO
HRhL2CO
L2RhCO
L2RhCO
O
CO
CH3CH2CH2CHO
1974 Kuntz - patent pro Rhone-Poulenc Ruhrchemie (nyní Hoechst) vývoj technologie - 300 000 t/rok n-butanalu (4 % iso-butanalu)
Rozpustnost 1100 g/l
Plynná fáze
Kapalina 1
Kapalina 2
Bifázová katalýza
Hydroformylace propylenu – proces 3. generace
Vodná bifázová katalýza
Substrát(y)
Substrát(y)
Fáze selektivněrozpuštěnéhokatalyzátoru
Fáze selektivněrozpuštěnéhokatalyzátoru
Produkt(y)
Produkt(y)
Před reakcí Po reakci
Ztráty rhodia: < 1 ppm
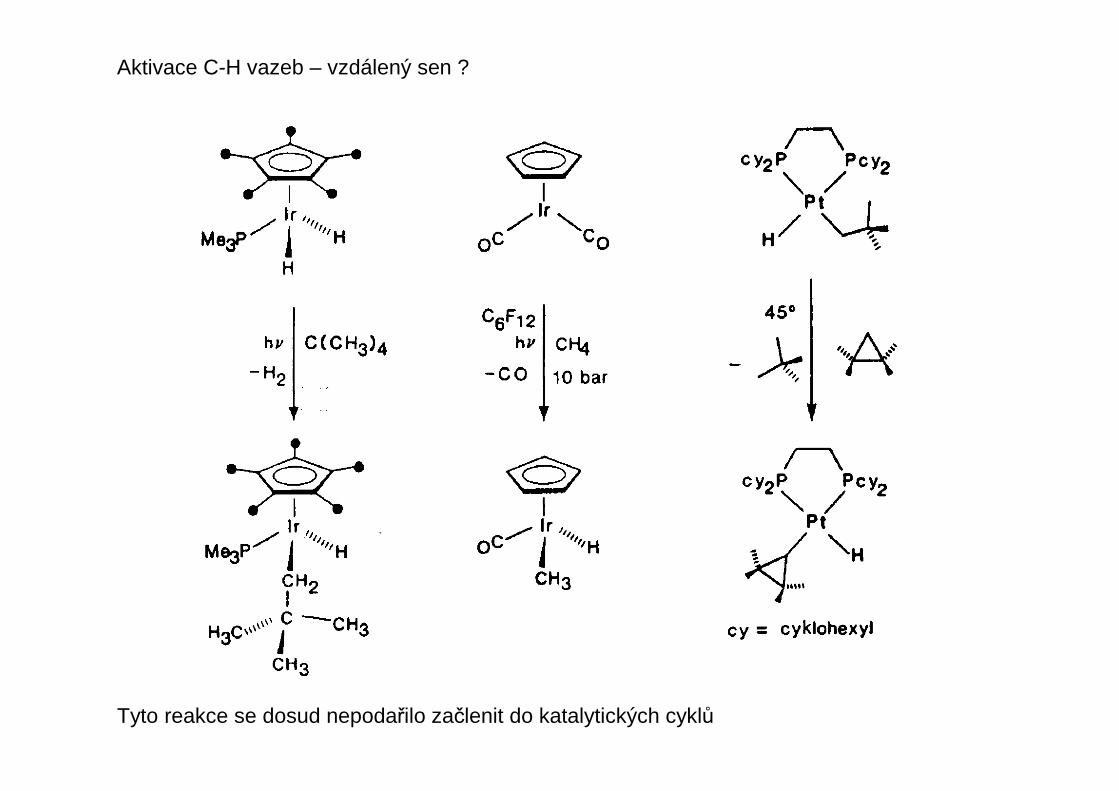
Aktivace C-H vazeb – vzdálený sen ?
Tyto reakce se dosud nepodařilo začlenit do katalytických cyklů
Literatura:
GREENWOOD, N. N., EARNSHAW, A. Chemie prvků I, II. Praha: Informatorium, 1993.
DVOŘÁK, D. Chemie organokovových sloučenin přechodných kovů. Praha: VŠCHT, 1994.
ELSCHENBROICH, C. Organometallics. 3rd ed. Wiley/VCH, Weinheim, 2006.
BLÁHA, K., FERLES, M., STANĚK, J. a kol.: Nomenklatura organické chemie. Praha: Academia, 1985.PANICO, R., POWELL, W. H., RICHER, J.-C. Průvodce názvoslovím organických sloučenin podle IUPAC, doporučení 1993. Praha: Academia, 2000.KLIKORKA, J., HANZLÍK, J. Názvosloví anorganické chemie. Praha: Academia, 1980.