111
LABORATORNÍ TECHNIKA PRO BIOCHEMIKY Petr Galuszka a Lenka Luhová Katedra biochemie Přírodovědecké fakulty Univerzity Palackého OLOMOUC 2005

LABORATORNÍ TECHNIKA PRO BIOCHEMIKY
Petr Galuszka a Lenka Luhová
Katedra biochemie Přírodovědecké fakulty Univerzity Palackého
OLOMOUC 2005

Recenzenti Prof. Ing. Jan Káš, DrSc. – Ústav biochemie a mikrobiologie FPBT VŠCHT Praha, Doc. RNDr. Petr Zbořil, CSc. – Katedra biochemie PřF MU Brno 2. vydání, Olomouc 2005 © Mgr. Petr Galuszka, Ph.D., RNDr. Lenka Luhová, Ph.D., 2005 Vydala Univerzita Palackého v Olomouci
2

Předmluva
Skripta byla sepsána jako přehledný soubor návodů pro absolvování předmětu Laboratorní technika. Praktické cvičení jsou v zimním semestru povinni navštěvovat posluchači prvních ročníků oboru biochemie a molekulární biologie na Přírodovědecké fakultě Univerzity Palackého.
Náplň cvičení byla koncipována tak, aby pokryla veškeré základní laboratorní techniky a postupy ze všech chemických a biologických oborů, se kterými se bude posluchač setkávat v praktických cvičeních během svého studia. Především je zaměřena na základní postupy uplatňující se v moderní biochemické laboratoři (práce s automatickou pipetou, spektrofotometrie, chromatografie). Velký důraz při sestavování úloh byl kladen na to, aby posluchači získali všechny základní návyky nutné pro bezpečnou a efektivní práci.
Skriptum je rozděleno na dvanáct samostatných laboratorních cvičení. Rozsahem odpovídá počtu hodin, které student musí absolvovat během semestru. Každé laboratorní cvičení obsahuje jeden nebo více úkolů, které jsou řešeny tak, aby je student stihl vypracovat během jednoho cvičení, tzn. zhruba za 4 hodiny čistého času. První dvě cvičení jsou úvodní a absolvují je všichni studenti společně. Dalších deset úloh je rozděleno podle náročnosti na dvě části a studenti se střídají v jejích vykonávání po týdnech. Prvních pět úloh v první půli semestru a zbylých pět náročnějších na konci semestru. Studenti většinou pracují v párech mohou však pracovat i jednotlivě, záleží na obsazenosti jednotlivého kurzu. V závěru každého cvičení je uvedena literatura, ze které autoři při sestavování čerpali, a několik otázek, na které studenti odpovídají na základě získaných dat či studia doporučené literatury. Vypracované odpovědi odevzdávají formou protokolu společně se stručným popisem a vyhodnocením cvičení.
Milou povinností autorů je poděkovat svým kolegům z katedry biochemie PřF UP za cenné rady a také posluchačům oboru biochemie v letech 2001-06 a 2002-07, kteří přímo za běhu cvičení vyjadřovali své připomínky a nejasnosti, na které byl brán zřetel při konečné podobě tohoto učebního textu.
V Olomouci, leden 2003
Autoři
3

PŘEHLED CVIČENÍ Z LABORATORNÍ TECHNIKY PRO BIOCHEMIKY
1. Úvodní hodina str.5
Laboratorní řád. Bezpečnost práce. Zdroje tepla v laboratoři. Měření teploty. Chemické sklo a laboratorní nádobí. Chemikálie a jejich uchovávání. Nebezpečné látky a manipulace s nimi.
2. Základní úkony v biochemické laboratoři str.16
Váhy a vážení. Měření objemů a pipetování. Příprava roztoků. Ředění kyselin a zásad. Pufry, jejich příprava a použití. Měření pH.
3. Úprava biologického materiálu, precipitační techniky, centrifugace, dialýza a ultra-filtrace str.26
Homogenizace semen ječmene, precipitace jejich proteinů síranem amonným s následnou dialýzou a ultrafiltrací na zařízení Amicon.
4. Extrakce, filtrace, sublimace a práce s vakuovou rotační odparkou str.35
Izolace kofeinu z čajového listí a jeho přečištění sublimací.
5. Spektrofotometrická měření str.42
Spektrofotometrické stanovení niklu v neznámém vzorku. Stanovení obsahu proteinů v biologickém materiálu metodou Bradfordové. Měření absorpčních spekter listových barviv. Stanovení koncentrace DNA.
6. Destilace str.50
Izolace silice z hřebíčku destilací s vodní parou.
7. Práce s plyny a sklem str.55
Určení procentuálního obsahu uhličitanu vápenatého ve vaječné skořápce jeho rozložením na oxid uhličitý. Práce se sklem a korkem.
8. Sušení, chlazení a lyofilizace str.63
Stanovení obsahu vody v modré skalici. Příprava bezvodého silikagelu. Teoretické seznámení s lyofilizátorem. Koncentrace roztoku proteinů pomocí gelové filtrace.
9. Krystalizace, promývání na filtru, dekantace a stanovení bodu tání str.73
Stanovení bodu tání (čistoty) izolovaného kofeinu. Izolace a krystalizace kyseliny citrónové z citronů. Rušená krystalizace technického síranu měďnatého.
10. Chromatografie I str.79
Sloupcová chromatografie na iontoměničích. Regenerace ionexu.
11. Chromatografie II str.86
Chromatografie karotenoidů na tenké vrstvě. Gelová chromatografie nízko- a vysokomolekulární látky.
12. Práce s mikroskopem str.94
Stavba rostlinné buňky v pokožce cibule. Plasmolýza, deplasmolýza, plazmoptýza. Histochemická reakce na vápník a na lignin.
13. Práce s mikroorganismy ve sterilním prostředí, izolace bakteriální DNA a agarosová elektroforesa DNA str.103
4

Laboratorní cvičení č. 1
ÚVODNÍ HODINA
LABORATORNÍ ŘÁD
1) Student je povinen se seznámit před zahájením práce v laboratoři s laboratorním řádem,
s bezpečnostními předpisy a s poskytováním první pomoci.
2) Student je povinen přicházet do laboratoře včas a řádně připraven. Musí mít provedeny potřebné výpočty, znát vlastnosti látek, se kterými bude pracovat apod. Před zahájením cvičení vyučující ověřuje znalosti studentů. Pokud student nemá dostatečné znalosti k řešení dané úlohy, cvičení vykoná v náhradním termínu.
3) Každá absence musí být omluvena. Má-li student vážné osobní důvody, pro které se nemůže zúčastnit cvičení, sdělí to vedoucímu předem. Každá zameškaná úloha musí být nahrazena. Na termínu náhradního cvičení se dohodne posluchač s vedoucím cvičení.
4) Při práci v laboratoři musí mít student pracovní plášť a vhodnou obuv.
5) Studenti pracují ve dvojících. Před zahájením práce zkontrolují podle přiloženého seznamu pracovní stůl a jeho vybavenost. Všechny závady zjištěné před zahájením práce nebo v jejím průběhu neprodleně hlásí vedoucímu cvičení.
6) Při práci je nutné postupovat přesně podle zadané úlohy a pokynů vyučujícího. Před používáním přístrojů se musí student nejprve seznámit s jejich obsluhou.
7) Průběh práce a dosažené výsledky si každý student zaznamenává do protokolárního sešitu. Po skončení cvičení předloží výsledky vedoucímu cvičení.
8) Následující cvičení odevzdává každý student vypracovaný protokol, který musí obsahovat: jméno studenta, studijní kombinaci, datum, název úlohy, stručný princip úlohy, stručný pracovní postup, výsledky, diskusi a závěr (tabulky, grafy).
9) Po skončení práce je student povinen dát své pracovní místo do pořádku, řádně umýt sklo a opláchnout je destilovanou vodou.
10) Student smí opustit laboratoř až po kontrole dosažených výsledků a stavu pracovního stolu vyučujícím.
11) Po skončení práce uzavřeme vodu a vypneme elektrické spotřebiče.
12) V laboratoři je zakázáno jíst, pít a kouřit.
13) Studenti jsou povinni dodržovat bezpečnostní předpisy.
14) Práce s jedovatými, těkavými a páchnoucími látkami se provádí pouze ve spuštěné digestoři.
15) Zvláštní opatrnosti je třeba dbát při manipulaci s otevřeným ohněm, hořlavinami, žíravinami a jedovatými látkami.
16) Případné závady, nedostatky, nehody nebo poranění je nutné ihned hlásit vyučujícímu a v případě potřeby poskytnout okamžitě první pomoc.
17) Práce v chemické laboratoři je zakázána těhotným ženám a matkám do konce 9. měsíce po porodu. Posluchačka je povinna vedoucímu cvičení okamžitě oznámit graviditu či nedávný porod.
5

BEZPEČNOST PRÁCE V CHEMICKÉ LABORATOŘI
1) Provádíme pouze práce podle pokynů vyučujícího a pracovního návodu.
2) Seznámíme se s rozmístěním hasících přístrojů a s únikovými východy z laboratoře.
3) V laboratoři nikdy nejíme, nepijeme a nekouříme. Po skončení práce si důkladně umyjeme ruce.
4) K jídlu a pití (mimo laboratoř) nikdy nepoužíváme chemické sklo.
5) Tašky a oblečení uložíme do skříní mimo laboratoř.
6) Při práci v laboratoři vždy nosíme pracovní plášť a vhodnou obuv.
7) Neprovádíme samovolné opravy nebo úpravy na elektrické instalaci a přístrojích.
8) Chemikálie nikdy nezkoušíme ústy a neinhalujeme výpary.
9) Nepipetujeme ústy.
10) Při práci se žíravinami a jinými nebezpečnými látkami si chráníme obličej a oči ochranným štítem, ruce gumovými rukavicemi.
11) Na pracovišti udržujeme pořádek a čistotu. Dbáme, abychom vnější stěny nádob nebo pracovní místo nepotřísnili chemikáliemi.
12) Koncentrované kyseliny a zásady ředíme tak, že kyselinu nebo zásadu lijeme tenkým proudem po tyčince do vody za současného míchání a chlazení.
13) Při provádění pokusů ve zkumavkách držíme ústí zkumavek odvrácené od obličeje (svého i spolupracovníků).
14) Při práci s hořlavinami nesmí být v blízkosti otevřený oheň. Při destilaci hořlavin je nezbytné z okolí odstranit zásobní láhve hořlavin a kontrolovat průtok vody chladičem. Hořlaviny nikdy nezahříváme přímým plamenem, používáme lázně nebo topná hnízda.
15) Zvýšenou pozornost věnujeme hlavně manipulaci s hořlavinami I. třídy, které mají teplotu vzplanutí do 21°C (aceton, ether, methanol, ethanol, benzín, benzen a toluen).
16) Pokud vypukne požár, je každý povinen pokusit se ho zdolat vlastními silami (hasicím přístrojem, improvizovanými hasicími prostředky). Je nutno dále vypnout elektrický proud a pokusit se odstranit z okolí požáru hořlavé látky (zejména kapaliny) a nádoby se stlačenými plyny. Nelze-li požár zvládnout vlastními silami, je nutné neprodleně volat požárníky (tel.číslo 150).
17) Střepy a jiné odpadky s ostrými hranami musí být odkládány do nádob zvlášť k tomu určených.
18) Zbytky jedů a organických rozpouštědel likvidujeme podle pokynů vyučujícího.
19) Při práci s etherem dbáme úzkostlivě na bezpečnostní opatření (možnost vznícení i od horkých součástí).
20) V případě nehody okamžitě informujeme vyučujícího a poskytneme první pomoc. Vedoucímu cvičení je třeba hlásit i každé nepatrném poranění, bolesti hlavy, hučení v uších apod. Ve všech případech je nutno sepsat protokol o poranění pro případ pozdějších komplikací.
6

PRVNÍ POMOC PŘI NEHODĚ
Při poleptání kůže silnou zásadou nebo kyselinou zasažené místo ihned důkladně omyjeme proudem vody. Při poleptání kyselinou provedeme neutralizaci roztokem hydrogenuhličitanu sodného (20 g.L-1), při poleptání zásadou zředěnou kyselinou octovou (5 g.L-1).
Při zasažení oka chemikálií ihned oko vypláchneme slabým proudem vody. V případě zasažení zásadou oko vypláchneme borovou vodou (roztok kyseliny borité 30 g.L-1). Jedná-li se o kyselinu, použijeme k výplachu roztok boraxu (20 g.L-1). V každém případě je nutné vyhledat očního lékaře.
Při poleptání sliznice v ústech provedeme důkladný výplach úst vodou a následně neutralizaci výplachem zředěnou kyselinou octovou (poleptání zásadou) nebo hydrogenuhličitanem sodným (poleptání kyselinou).
Při požití louhu se doporučuje pít zředěnou kyselinu octovou (0,5 - 2,0 g.L-1), při požití kyseliny pijeme suspenzi oxidu hořečnatého nebo hydroxidu hlinitého ve vodě. Po požití jedů je charakter první pomoci specifický podle druhu otravy, doporučuje se vypít aspoň 0,5 l vody a vyvolat zvracení. Je vždy nutné vyhledat odborné lékařské ošetření.
Hořící oděv hasíme přikrývkou nebo sprchovou vodou. Při likvidaci větších plamenů použijeme hasící přístroje. Při malých popáleninách ošetříme postižené místo mastí na spáleniny a zakryjeme sterilním obvazem. Větší popáleniny ošetří lékař.
Při pořezání sklem odstraníme z povrchové rány sklo, okolí otřeme zředěným peroxidem vodíku (3%) a ovážeme sterilním obvazem. Větší zranění ošetří lékař.
Nebezpečné látky v chemické laboratoři:
Amoniak je značně těkavý. Páry leptají sliznice a ve vysokých koncentracích může působit smrtelně. Postižený potřebuje naprostý klid a pobyt na čerstvém vzduchu. Při nadýchání či požití je nutné podávat hodně vody s octem nebo citronem, později olivový olej s kousky ledu. S amoniakem pracujeme vždy v digestoři.
Chlor, brom silně leptají dýchací orgány. Jejich účinky se projevují až po několika hodinách (kašel, dušení). Postiženého přeneseme na čerstvý vzduch a zajistíme klid. Nenutíme ho dýchat zhluboka a nikdy nezavádíme umělé dýchání. Je možné vdechovat vodní páru s amoniakem nebo ethanolem, či 2% roztokem sody. Asanace rozlitého bromu se provádí thiosíranem.
Kyanovodík a kyanidy jsou prudce jedovaté látky mandlového zápachu. Páry kyanovodíku způsobují i v nepatrných koncentracích okamžitou smrt. Plyn proniká i pokožkou. Při práci s kyanovodíkem používáme vždy plynovou masku. Při otravě nutná okamžitá lékařská pomoc. Dlouhodobě zavádíme umělé dýchaní, popř. zabezpečíme vdechování kyslíku. Kyanidy působí jako inhibitory dýchacího řetězce mitochondrií. Smrtelná dávka je zhruba 200 mg. Při otravě je potřeba okamžitě vypláchnout žaludek, popř. vyvolat zvracení. Podáváme zředěný peroxid vodíku. Injekčně se podává methylenová modř a thiosíran. Páry kyanovodíku se uvolňují v kyselém prostředí z kyanidů!
Oxid uhelnatý je prudce jedovatý plyn, který nelze zjistit čichem. Vytěsňuje kyslík z vazebného místa na hemoglobinu. Již po krátkém nadýchání otupuje a způsobuje smrt zadušením. Postiženého vyneseme na čerstvý vzduch a zavádíme umělé dýchání s inhalaci kyslíku.
Nitrózní plyny mohou způsobit i při vdechnutí malého množství smrt. Postiženého je třeba nechat vdechovat volně kyslík a přičichávat amoniak. Nezavádíme umělé dýchaní a poskytneme absolutní klid.
Oxid siřičitý vyvolává při vdechování křečovitý kašel. Postiženého je třeba přenést na čerstvý vzduch, popř. jej nechat vdechovat páry etanolu nebo kyslíku.
7

Sulfan je prudce jedovatý plyn. Při nadýchání je potřeba postiženého ihned vynést na čerstvý vzduch. Dojde-li k otravě a ztížení dýchání, je nutné okamžitě inhalovat kyslík a zavádět umělé dýchání i při zdánlivé smrti.
Rtuť je jedovatá nejen ve svých sloučeninách, ale i v parách a prachu. Při otravě je potřeba podávat prostředky na zvracení (mýdlová voda) a vypláchnout žaludek. Pak podávat bílek v mléce, vodnou suspenzi hořčíku s mandlovým olejem, roztok taninu nebo železný prach. Při rozlití asanovat malé kapičky sirným květem nebo posbírat mosazným plíškem. Při rozbití rtuťového teploměru je nutno vždy řádně odstranit veškerou rtuť z prostředí, jelikož ta pak může v laboratoři způsobovat dlouhodobé otravy.
Olovo je nebezpečné v parách a jeho sloučeniny způsobují dlouhodobě chronickou otravu, tzv. saturnismus. Po požití je třeba podávat velké množství roztoku MgSO4, vyvolat zvracení a pak podávat mléko a vodu s bílkem. Po práci s kovovým olovem si vždy řádně opláchněte ruce.
Methanol působí škodlivě jednak vdechováním par, jednak požitím. V první fázi vyvolá opojení později křeče. Větší dávky způsobují trvalé oslepnutí a smrt. Na rozdíl od etanolu se velice dobře vstřebává i kůži. Při práci s metanolem proto vždy pracujeme v ochranných pomůckách. Při požití metanolu se jako antidotum podává etanol, který je daleko lepším substrátem lidské alkoholdehydrogenasy a zabraňuje tak tvorbě toxického formaldehydu z metanolu.
Benzen, toluen, naftalen působí škodlivě na červené krvinky. Při požití vyvolává zvracení. Je nutný klid, popř. umělé dýchaní.
Fenol je značně toxická látka leptající kůži, kterou se rychle vstřebává a způsobuje celkovou intoxikaci. Potřísněnou kůži omýváme 25% alkoholem nebo glycerolem. Při práci s fenolem se vždy chráníme rukavicemi.
Zvláště nebezpečnou skupinou látek, se kterými se můžete v chemické laboratoři setkat, jsou látky mutagenní a karcinogenní. Jejichž nebezpečnost tkví v tom, že nezpůsobují akutní otravy, ale dlouhodobým působením mohou vyvolat nádorové onemocnění. Při práci s těmito látkami pracujeme vždy dle daným předpisů a chráníme se ochrannými pomůckami. Mezi karcinogeny, které se často používají v biochemické laboratoři patří: benzen, styren, chloroform, ethidium bromid a akrylamid.
Při práci s infekčním materiálem je nutno zachovávat všechna hygienicko-preventivní opatření, abychom nenakazili sebe ani své okolí. Za potenciálně infekční materiál je nutno považovat všechny tělesné tekutiny (moč, sliny, krev) a živočišné tkáně, případně extrakty z nich připravené.
LABORATORNÍ SKLO A PORCELÁN
Mezi laboratorní sklo řadíme veškerý skleněný materiál, se kterým se setkáváme v chemické laboratoři. Laboratorní sklo má vysokou odolnost k minerálním kyselinám a zásadám. Z fyzikálních vlastností skla je nejdůležitější jeho tepelná roztažnost. Obecně mají skla měknoucí při vyšších teplotách větší odolnost proti náhlým tepelným změnám. Setkáváme se především se třemi druhy skla:
1) Sklo měkké – sodno-draselno-vápenaté se používá na výrobu nádobí, které není vystavováno tepelnému namáhání. Má velký koeficient roztažnosti, takže nesnese kolísání teploty. Musí se zahřívat nebo ochlazovat velmi opatrně. Má nízký bod tání a proto se dá snadno roztavit v plameni nad kahanem. Změklé sklo se pak dá lehce opracovávat.
2) Sklo tvrdé - borosilikátové slouží na výrobu skleněného nádobí, které můžeme zahřívat nad plamenem. Zhotovuje se z něj veškeré varné a odměrné sklo. Borosilikátové sklo má nazelenalou barvu a vyznačuje se odolností proti praskání, vysokým bodem tání a vysokou chemickou odolností. Nejběžnější obchodní značky jsou Sial, Simax, Duran či Pyrex.
8

3) Sklo křemenné – se vyznačuje vysokou chemickou a tepelnou odolnosti. Je však velice křehké a využívá se pouze ke zhotovování speciálních nádob a zařízení, např. kyvet pro spektrofotometrii (propouští UV záření).
Obecnou vlastností skla je jeho křehkost. Zbytečnému praskání zamezíme řádným uchycením do svorek a lapáků s čelistmi s korkovou výztuží, popř. s navlečenými gumovými hadičkami. Při práci s agresivními látkami či ve vakuu není vhodné používat korek a gumové hadice. Pracujeme pak se zábrusovým sklem, které se dá vzájemně stavebnicově propojovat. Zábrusové baňky, chladiče, teploměry, zátky atd. jsou normalizované. Nejčastější rozměry zábrusů jsou NZ 14,5/15; NZ 29/32; NZ 40/45 (čísla znamenají průměr zábrusu v mm ve zúžené a širší části). Práce se zábrusy je rychlá a pohodlná. Zábrusy je však třeba vždy řádně promazat silikonovou vaselinou, aby nedocházelo k jejich „zatuhnutí“.
Vedle skleněného nádobí používáme v chemické laboratoři velice často též nádobí a pomůcek z porcelánu (třecí a odpařovací misky, žíhací kelímky, váženky, lžičky, Bűchnerovy nálevky aj.). Tvrdý chemický porcelán má vysokou mechanickou a chemickou odolnost. Je citlivý na údery a lehce se tříští hlavně při prudkých změnách teploty. Je stálý proti vzdušnému kyslíku i za žáru, vodu povrchově neváže a je v ní i za vysokých teplot nerozpustný. Chemickým činidlům vzdoruje asi stejně dobře jako chemické sklo. Chemický porcelán může být drsný či glazurovaný. Glazura jeho vlastnosti nijak výrazně neovlivňuje.
Chemické nádobí čistíme ihned po práci, dokud nečistoty a zbytky chemikálií na stěnách ještě nezaschly. Zpravidla si vystačíme s postupy známými z domácnosti, tj. použití saponátového prostředku a důkladné omytí vodou. V laboratoři však nezapomene na poslední krok, kterým je řádné vypláchnutí destilovanou vodou. Pokud na stěnách ulpěly kousky ve vodě nerozpustných nečistot použijeme mechanických pomůcek jako jsou kartáče, útržky filtračního papíru nebo jemný písek. Toto mechanické čištění však nesmí sklo poškrábat, i nepatrné škrábnutí může způsobit při zahřívání prasknutí skla. Když ani mechanické čištění nevede k odstranění nečistot, přichází na řadu chemické čištění. Použijeme rozpouštědlo, které čištěný materiál nekoroduje a ve kterém je nečistota rozpustná. Nejčastěji se využívají v laboratoři dostupné minerální kyseliny. Při této činnosti dbáme na dodržování bezpečnostních předpisů a používáme ochranné pomůcky. Vysokou čistící schopnost má tzv. kyselina chromsírová, což je dichroman sodný rozpuštěný v koncentrované kyselině sírové. Sklo se do této směsi namočí přes noc a ráno se pak důkladně opláchne v roztoku detergentu, pod tekoucí vodou a nakonec ve vodě destilované. Chromsírová směs se po čase vyčerpá, což se projeví zeleným zbarvením (redukce dichromanu na ion chromitý). Taková směs je pak málo účinná a musí se připravit nová.
Zvláštní nároky jsou kladeny na sklo používané v molekulárně biologické laboratoři, kde se velice často pracuje s bakteriálními kmeny a jinými mikroorganismy a je velké riziko bakteriální kontaminace z okolí. Proto se snažíme používat pro manipulaci s těmito organismy pouze sterilní plasty na jedno použití. Pokud je nutno použít skleněné či jiné dražší nádobí, musí se předem sterilizovat ve speciálních tlakových nádobách (autoklávy) a následně vysušit v sušárně na 105°C. Zvláštní péče je pak věnována sklu, které se používá pro manipulaci s RNA. Hrozí totiž nebezpečí kontaminace ribonukleasami, což jsou degradační enzymy RNA, které jsou velice stabilní a odolávají i běžné sterilizaci. Sklo se proto před sterilizaci namáčí do roztoku jejich inhibitoru diethyl pyrokarbonátu.
CHEMICKÉ NÁDOBÍ A APARATURY
Mezi základní vybavení každé chemické laboratoře patří baňky a kádinky. Kádinky mají rovné dno a na horním okraji mohou mít zobáček. Mohou být i kalibrovány, ale tato kalibrace slouží pouze k orientačním účelům, rozhodně podle ní nelze odměřovat přesné objemy. Baňky mají tvar členitější, rozdělený na vlastní baňku a hrdlo. Dno mohou mít jak rovné, tak kulaté, ty se pak používají hlavně pro práci za sníženého tlaku. Baňky a kádinky jsou převážně tenkostěnné,
9

1 2 3 4 5 6
7 8 9 10 11 12
13 14 15 16 17 18 19 20
21 22 23 24 25 26 27 28
Obrázek 1: Laboratorní nádobí: 1/ kádinka s výlevkou, 2/ kádinka bez výlevky, 3/destilační baňka se zábrusem a kulatým dnem, 4/ destilační baňka se zábrusem a plochým dnem, 5/ hruškovitá baňka, 6/ Erlenmeyerova baňka se zábrusem, 7/ baňka trojhrdlá, 8/ titrační baňka, 9/ frakční baňka, 10/ baňka odsávací, 11/ odměrná baňka, 12/ dělicí nálevka kulovitá, 13/ dělící nálevka hruškovitá, 14/ nálevka s dlouhým stonkem, 15/ nálevka s krátkým stonkem, 16/ násypka, 17/ Büchnerova nálevka, 18/ skleněná frita pro vakuovou filtraci, 19/ promývačka, 20/ láhev se šroubovacím uzávěrem, 21/ úzkohrdlá zábrusová láhev, 22/ zkumavka, 23/ zkumavka se šroubovacím uzávěrem, 24/ centrifugační kyveta, 25/ Petriho miska, 26/ krystalizační miska, 27/ odpařovací miska, 28/ hodinové sklo.
10

jednou z vyjímek je silnostěnná baňka odsávací. Slouží k filtraci za sníženého tlaku a je určená pouze k tomuto účelu, v žádném případě v ní nelze provádět nějaké chemické reakce, plnit horkými kapalinami nebo zahřívat. Baňka konického tvaru se nazývá Erlenmayerova. Baňku s postranním tubusem nazýváme frakční.
Slovem nálevka označujeme v laboratoři větší počet pomůcek. Obyčejné nálevky slouží k přelévání kapalin a jednoduchým filtracím. Pro urychlení filtrace se používají nálevky s žebrovaným vnitřním povrchem nebo dlouhým a úzkým stonkem. Tzv. dělící nálevky se používají k řadě procesů, jako je extrakce, sušení, přikapávání atd. Velmi často používanou je, z porcelánu vyrobená, Büchnerova nálevka mající dírkované dno, na které se při filtraci vkládá kolečko filtračního papíru. Podobně vypadají i skleněné frity, které mají místo dírkovaného dna sintrované sklo propouštějící rozpouštědla. Zábrusové láhve na čištění plynů se nazývají promývačky. Zásobní láhve na chemikálie se dělí podle typu hrdla na reagenční láhve se šroubovacím víkem, sloužící především k uchovávání kapalin a širokohrdlé se zábrusovou zátkou tzv. prachovnice, ve kterých přechováváme pevné látky. K přechovávání malých množství pevných látek používáme lékovky. Protáhlá neuzavíratelná skleněná nádobka se nazývá zkumavka, slouží k provádění chemických reakcí v malých objemech. V poslední době jsou velmi populární plastové zkumavky se šroubovacím uzávěrem. Nádoby podobného tvaru, většinou také šroubovací, používáme k centrifugaci a nazýváme je kyvety. Bývají zhotovovány většinou z tvrzených plastů. Hodinová skla slouží k přikrývání nádob, k sušení, vážení a přenášení vzorků pevných látek. Analogicky můžeme využívat i dvoudílné Petriho misky, jež se hlavně používají pro kultivace mikroorganismů. Odpařovací misky bývají většinou vyrobeny z porcelánu. Nejsou vhodné k přímému zahřívání v plameni stejně jako misky krystalizační. Pro odpařování roztoku používáme vždy vodní lázeň. K drcení a roztírání pevných vzorků slouží třecí misky s tloučkem. Jediným porcelánovým nádobím, které snáší přímý oheň, jsou žíhací kelímky.
Chemické sklo většinou nepoužíváme samostatně, ale sestavujeme ho do různých aparatur, např. aparatura na destilaci, sublimaci apod. Aparatury sestavujeme na kovových stojanech nebo kovových klecích. Na svislé tyče stojanů a klecí připevňujeme kruhy, svorky nebo držáky pomocí svorek (obr. 2). Na kovové kruhy zpravidla pokládáme azbestové síťky, na které stavíme kádinky v případě jejich zahřívání kahanem. Kruhy pro zahřívání nikdy nezaměňujeme s kruhy filtračními, které jsou vyloženy dřevěnou nebo umělohmotnou vložkou a slouží k držení nálevky při filtraci nebo dělící nálevky při dělení kapalin. Kruh na stojanu se často nahrazuje trojnožkou. K upevňování skleněných součástí malého průměru používáme tzv. klemy, což jsou držáky s malým obvodem ramen. Pro uchycování chladičů pak používáme chladičové držáky s velkým rozpětím ramen. Klemy a držáky se nesmějí nikdy dotýkat skleněných částí aparatury přímo kovem. Bývají vyloženy korkem nebo potaženy gumovou hadicí. Při sestavování jakékoli aparatury musíme dbát na to, aby v aparatuře nenastalo pnutí, které by při zahřátí vedlo k prasknutí některé ze skleněných součástí. Při sestavování aparatury ze zábrusového skla nikdy neopomeneme zábrusy řádně promazat vazelinou.
Obrázek 2: Sestavování aparatur. A – nesprávné uchycení křížové spojky, B – správné uchycení, C – chladičový držák, D – klema.
11

ZDROJE TEPLA V CHEMICKÉ LABORATOŘI
V laboratořích, do kterých je zaveden plyn, nám jako zdroj pro zahřívání může sloužit plynový kahan. Kahan je jednoduché zařízení sloužící k tvorbě směsi vzduchu a plynu, která se spaluje a vytváří na konci trubice plamen. Podle způsobu, jakým je k plynu přiváděn a mísen vzduch, rozlišujeme tři typy kahanů: Bunsenův, Tecluho a Mékerův. Pokud je do plynu přiváděno dostatečné množství vzduchu hoří nesvítivým plamenem, který je výhřevnější než plamen svítivý, jež získáme zamezením přívodu vzduchu. Z toho plyne pravidlo, že čím více vzduchu smísíme s plynem, tím výhřevnější plamen získáme. U Bunsenova a Mékerého kahanu se přívod vzduchu reguluje otáčením pohyblivého prstence, který odkrývá nebo zakrývá otvory v těle kahanu. Kahany zapalujeme vždy při uzavřeném přívodu vzduchu. V laboratoři často narazíme i na kahan lihový, který má daleko nižší výhřevnost než kahany plynové. Výhodou však je jeho velikost a jednoduchost. Lze ho použít všude tam, kde potřebujeme vzorky či chemické reakce jen zahřát. Používá se hlavně při zkumavkových reakcích a pro sterilizaci materiálu při práci s mikroorganismy.
V současné době se však jako hlavní zdroje tepla v chemických laboratořích využívají elektrické vařiče a topná hnízda. Na elektrický vařič pokládáme vždy azbestovou síťku. Topná hnízda používáme hlavně na zahřívání destilačních baněk. Jde o elektrický vařič, u kterého je elektricky vyhřívaná speciální tkanina vytvarovaná do polokoule tak, aby obalila destilační baňku. Existuje několik typů topných hnízd podle velikosti zahřívané baňky. U topného hnízda lze kromě regulace příkonu většinou regulovat i to, zda je vyhřívaná pouze spodní část baňky, nebo baňka celá. Při práci s topnými hnízdy je třeba hlavně dávat pozor, aby do hnízda nevnikla kapalina (voda). Dojde-li k tomu, neprodleně přístroj vypneme.
Protože každé přímé zahřívání klade vysoké nároky na materiál nádob, je výhodné předávat teplo zdroje prostřednictvím různých lázní. Podle materiálu, který tvoří podstatu lázně je možné dělení na vzdušné, vodní, olejové, pískové, kovové a solné. Vzdušné lázně se moc často nepoužívají vzhledem k tomu, že vzduch je ze všech materiálů nejméně vodivý. Nejjednodušší formou vzdušné lázně je prázdná kovová nádoba zahřívána kahanem nebo vařičem. Nejčastěji používáná je lázeň vodní, která je vhodná k zahřívání látek až do bodu varu vody a k destilaci kapalin vroucích přibližně do 80°C. Nejjednodušší vodní lázní je obyčejný hrnec, který je vyhříván elektrickým vařičem. V laboratoři se lze setkat i se speciálními vodními lázněmi, které mají různě nastavitelné otvory pro zahřívání odlišných velikostí baněk a odpařovacích misek. Pro zahřívání nad teplotu varu vody se nejčastěji používají lázně olejové. Náplní může být buď olej minerální, který lze použít do teploty 250°C, nebo silikonový použitelný až do teplot okolo 400°C. Vzhledem k teplotní roztažnosti je nutno olejem naplnit nádobu sloužící jako obal lázně jen asi do poloviny, jinak by při zvýšení teploty mohl olej přetéci. Při zahřívání je nutno do olejové lázně vždy vložit teploměr a průběžně kontrolovat teplotu, aby nedošlo k jejímu přehřátí. Obvykle platí, že teplota lázně, má být o 20-30°C vyšší než žádaná teplota reakční směsi. Je důležité, aby se do olejové lázně nedostala voda. Při teplotě nad 100°C by pak došlo k prskání a pěnění oleje, který by mohl způsobit popáleniny osobě pracující s lázní nebo požár. Solné a kovové lázně slouží k zahřívání nad 300°C. Používá se směs několika solí, např. dusičnan sodný a draselný o bodu tání 219°C a nízkotající slitiny, jako je Woodoův kov s bodem tání 65°C. Vždy je však důležité vyjmout baňku před ztuhnutím lázně. Výhodou těchto lázní je vysoká tepelná vodivost použitých materiálů. Od používání písečných lázni se už v podstatě v dnešní době ustoupilo.
K žíhání a tavení většího množství látek nám slouží elektrické odporové pece. Pro syntézy v proudu plynu nebo reakce ve vakuu užíváme pece trubkové, pro žíhání v kelímcích nám slouží kelímkové pícky a pro tavení většího množství látek pece muflové.
12

VAKUUM A JEHO ZDROJE
V laboratorní praxi je velmi často třeba pracovat za sníženého tlaku. Sníženého tlaku - vakua se využívá hlavně při destilaci, filtraci či sušení, kde výrazně ovlivňuje těkavost látek. Vakuum je stav uzavřeného prostoru, ve kterém je tlak plynu nebo páry nižší než tlak atmosférický okolního prostředí.
Zařízení vytvářející vakuum nazýváme vývěvou. V laboratoři se můžeme setkat s vývěvami několika typů, které se liší tlakem, proti kterému čerpají, mezní hodnotou vakua a sacím výkonem. Nejjednodušším typem vývěvy je vývěva vodní. Jedná se o zúženou trubici, kterou tryská proud vody. V okolí ústí trysky je vzduch strháván ve směru proudu vody a je zde napojena odvodná hadice, která se druhým koncem napojuje na evakuovanou aparaturu. Vodní vývěva pracuje tedy přímo proti atmosférickému tlaku. Mezní tlak vakua je dán tenzí vodní páry (pro vodu o teplotě 10°C to je 1333 Pa). Mezi vodní vývěvu a aparaturu vždy zařazujeme pojistnou nádobu (promývačku), aby nedošlo při poklesu tlaku vody v potrubí ke vniknutí vody do aparatury. Vývěvy dále mohou být rotační olejové nebo mechanické, jejich konstrukce je již složitější. Základním stavebním prvkem rotační olejové vývěvy je rotor se šoupátkem, které rozděluje prostor mezi rotorem a pláštěm na dvě části. Otáčením rotoru se jeden prostor zvětšuje a nasává vzduch a druhý prostor se zároveň zmenšuje a vzduch je vytlačován za součinnosti ventilu z vývěvy. Hlavním pravidlem při práci s vývěvou je, že vždy po skončení práce nejdříve znovu zavzdušníme aparaturu a až poté vypneme zdroj vakua. Zabráníme tím vniknutí vody do aparatury, či nasátí agresivních látek do vývěvy a její poškození.
MĚŘENÍ TEPLOTY
Pro měření teploty byla přijata mezinárodní teplotní stupnice, definovaná pomocí bodu tuhnutí vody a varu vody. Další pevné body mezinárodní stupnice jsou: bod varu síry – 444,6°C, bod tání stříbra – 960,8°C a bod tání zlata – 1062,4°C. Všechny tyto teploty jsou definovány za atmosférického tlaku jedné atmosféry. Teplotu měříme teploměry, které mohou být trojího typu: dilatační, odporové a termočlánky.
Dilatační teploměry využívají roztažnosti kapalných, popřípadě pevných či plynných látek. Nejčastěji používané jsou teploměry rtuťové. Dovolují měřit teplotu od –38,9°C do 650°C. Pro měření nízkých teplot se teploměry plní toluenem (-80°C až 100°C), etanolem (-100°C až 70°C), petroléterem (-150°C až 120°C) či pentanem (-190°C až 20°C). Pro vyšší teploty se pak používá jako náplň kapalné galium (do 1000°C) a cín (do 1500°C).
Odporové teploměry využívají závislosti odporu vodiče na teplotě. Těmito teploměry lze měřit teplotu s přesnosti na tisíciny stupně.
Termočlánky jsou založeny na termoefektu – dva dráty ze dvou různých kovů spojené na obou koncích dávají vznik elektrického proudu, jsou-li oba spoje udržovány na rozdílné teplotě. Příkladem nejčastěji používaných termočlánku je: měď – konstantan (do 600°C), železo – konstantan (do 900°C), platina – platinarhodium ( do 1600°C).
VODA V BIOCHEMICKÉ LABORATOŘI
Vzhledem k tomu, že obyčejná tekoucí voda obsahuje značné množství solí, je pro jakékoli pokusy v biochemické laboratoři nepoužitelná. Používáme ji pouze tehdy, když nepřichází do styku s ostatními reagenciemi, tedy jako rozpouštědlo, např. pro chlazení.
Základním způsobem úpravy vody je destilace. Každá biochemická laboratoř bývá vybavena vlastní destilační aparaturou. Destilovaná voda postačí pro většinu biochemických operací, ale pro některé speciální účely nemusí být její čistota vhodná. Obsahuje především některé kationty, které se do ní dostávají ze součástí destilační aparatury, skla a elektrod. Její kvalitu lze zvýšit
13

opakovanou destilací tzv. redestilací, která se provádí ve speciální aparatuře z křemenného skla neuvolňující kationty.
Pro speciální účely, např. v laboratoři molekulární biologie, kde vadí i sebemenší kontaminace vody, se používá voda deionizovaná, která se ještě následně sterilizuje v autoklávu. Příprava deionizované vody je založená na kombinaci několika separačních metod vedoucí k odstranění jednotlivých skupin kontaminantů. Souprava je většinou složená z dílčích separátorů odstraňujících hrubší nečistoty (filtry), ionty (iontoměniče), nepolární látky (adsorbent) a jako poslední bývá zapojen membránový filtr. Některé aparatury pracují i na principu reverzní osmózy. Jednotlivé filtry je však nutno po několika měsících provozu vyměňovat a příprava deionizované vody není proto levnou záležitostí.
Hlavním kritériem čistoty vody je její specifická vodivost. U běžně destilované vody se pohybuje okolo hodnoty 10 µS.cm-1. Deionizovaná voda pak mívá tuto hodnotu ještě o řád nižší. Připravenou destilovanou či deionizovanou vodu skladujeme převážně v plastových nádobách. Skleněné nádoby nejsou pro dlouhodobější uskladnění vhodné, jelikož se do vody zpětně uvolňují některé kationty. A pokud to podmínky dovolují, skladujeme vodu v chladu a ve tmě, čímž zabráníme možné kontaminaci autotrofními mikroorganismy.
CHEMIKÁLIE A JEJICH UCHOVÁVÁNÍ
Jako výchozí látky pro práci v laboratoři používáme většinou průmyslově vyráběné chemikálie opatřené výrobcem štítkem udávající nám základní informace o dané chemikálii.
Chemikálie jsou dodávány výrobcem ve vhodném obalu, proto se snažíme je dále nerozdělovat do obalů jiných. Pokud je to nutné, vždy použijeme vhodný obal a dodržujeme základní pravidla. Látky kapalné uchováváme v láhvích s dvojím uzávěrem. Látky hygroskopické chráníme proti vzdušné vlhkosti utěsněním víčka, např. parafilmem. Hydroxidy a jejich roztoky neskladujeme v zábrusových lahvích. Látky citlivé na světlo skladujeme v tmavých nádobách a ve tmě. Skladování chemikálií v laboratoři má svůj pevný řád, který vychází z bezpečnostních pravidel, a proto vždy používanou chemikálii vracíme na původní místo. Usnadníme tím i práci kolegům, kteří budou s danou chemikálii následně pracovat. Důraz je kladen především na hořlaviny a látky výbušné, které nikdy nesmí být uskladněny pohromadě ve větším množství. Jedy skladujeme ve speciálních železných skříních pod zámkem. Pozor dáváme taky na uskladnění těkavých látek v uzavřených prostorách (lednice), skladujeme je tak, aby nedocházelo k uvolňování jejích par.
Na štítku chemikálie je především její název, sumární vzorec, množství, čistota a molekulová hmotnost. Dále mohou být uvedeny některé důležité fyzikální vlastnosti a bezpečnostní informace (obr. 3 a 4). U pevných látek bývá uvedená forma např. krystalický (crystalisatum), bezvodý (anhydricum), práškový (pulveratum). Podle rostoucí čistoty (tzn. klesajícího obsahu nečistot) chemikálie dělíme na:
a) technické chemikálie
- surové (crudum) - technické (technicum) - čištěné (purum)
b) čisté chemikálie
- čisté (purissimum) - pro analýzu (p.a., per analysi) - chemický čisté (purissimum speciale)
14

Chemikálie o vyšší čistotě jsou pak označeny přímo účelem, pro který slouží, např. pro UV spektrofotometrii, molekulární biologii atd. S čistotou chemikálií však prudce roste jejich cena, proto chemikálie o vysoké čistotě používáme jen pro tyto speciální účely a je-li to nezbytně nutné.
Obrázek 3: Příklad štítku chemikálie firmy Sigma. A – název chemikálie, B – katalogové číslo, C – informace o čistotě a fyzikální vlastnosti, D – doporučená manipulace a podmínky skladování, E – údaje o riziku, F – minimální obsah, I – piktogram označující rizika, L – sumární vzorec a molekulová hmotnost.
Obrázek 4: Piktogramy používané pro označení nebezpečných chemikálií.
15

Laboratorní cvičení č. 2
ZÁKLADNÍ ÚKONY V BIOCHEMICKÉ LABORATOŘI
VÁHY A VÁŽENÍ
Ke stanovení váhového množství látky užíváme v chemické laboratoři váhy. Princip vážení je znám po staletí: jde o srovnávací metodu, kdy se srovnává neznámá hmotnost nějakého předmětu (navažovací lodička, mikrozkumavka se vzorkem) se známou hmotností standardu (závaží). Mechanické zařízení řešilo toto srovnání pomocí docílení rovnováhy na páce a mělo tvar známých miskových vah, které vešly v obecné povědomí i jako symbol používaný pro zobrazování spravedlnosti. Až do nedávna byly i nejpřesnější analytické váhy postaveny na stejném principu a vážení obsahovalo postupné přidávání závaží různé (i velmi malé) velikosti. Takový postup byl samozřejmě zejména pro začátečníky velmi zdlouhavý a možnosti chyb četné.
V současné době patří všechny formy dvoumiskových vah historii a v laboratoři se setkáváme výhradně s vahami jednomiskovými používající promítací stupnice na stínítko a tedy zapojených do elektrické sítě. Existují dva rozdílné typy vah lišící se váživostí tzn. maximální hmotností, kterou můžeme vážit, a přesností, se kterou na nich můžeme hmotnost stanovit:
1) Váhy technické mají podle provedení váživost od 100 g do několika kilogramů. Přesnost nebývá větší než 5.10-2 g.
2) Váhy analytické mají váživost obvykle 100 g a přesnost v řádu 10-4 g.
K orientačnímu zjištění hmotnosti předmětů, které budeme dále vážit přesněji, a ke zjišťování výtěžků čistících operací používáme tzv. předvážky. Slouží k vážení předmětů do 200 g s přesností ± 0,1 g. V dnešní době se už používají převážně elektronické typy s digitálním displayem. Jejich forem je řada a procházejí stále vývojem od jednodušších s mechanicko-elektrickým snímačem hmotností přes snímače, kdy je měřen elektrický signál potřebný k vrácení misky po zatížení do nulové polohy pomocí servomotorku, až k snímačům tenzometrickým.
V našem praktiku nalezneme dva typy analytických vah: robusnější mechanické a moderní elektronické. K účelům cvičení z laboratorní techniky budeme používat především mechanické jednomiskové váhy. Jejich princip je znázorněn na obrázku 5. Je zřejmé, že jde vlastně o klasické jednoramenné váhy pouze upravené pro co nejsnazší obsluhu. Podstatou jejich funkce je rovnováha na vahadlu, které je zpravidla z lehké slitiny. Aby pohyb vahadla mohl být co nejjemnější, je jeho osou břit, tj. trojboký hranol z velmi tvrdého materiálu (achát) spočívající hranou na vyleštěné podložce. Je patrné, že jakékoli prudké pohyby vahadla by v tomto uspořádání kladly na břit extrémní nároky a velice brzy by byl břit opotřebován. Proto je v klidovém stavu břit z podložky zvednut a mechanismus spočívá v pevných lůžkách. Tomuto stavu říkáme aretace. Je pravidlem, že veškeré manipulace, jako je vkládání vzorku do vah či přidávání nebo ubírání závaží, provádíme výhradně na zaaretovaných vahách. Odaretování, tj. spuštění břitu na podložku, lze provést pouze v situaci, kdy očekáváme na vahadle rovnováhu. Tento úkon je nutné provádět pomalu a jemně. Váhy jsou konstruovány tak, že při odaretování se zapojí elektrický proud a rozsvítí se stinítko, na které je promítána stupnice.
16

Obrázek 5: Schéma jednomiskových vah (B – břit, V – vahadlo, Z – závaží, M – miska, P – proti-závaží, S – stupnice).
Z obrázku 5 je zřejmé, že na jedné straně vahadla je upevněná miska a závěs, na kterém jsou ovladatelná závaží až do hodnoty váživosti, na straně druhé pak pevné protizávaží. Je-li miska vah prázdná, je miska se závěsem a závažími právě vyvážená protizávažím a po odaretování se na světelné stupnici promítne nula. Pokud tomu tak není, jde nejčastěji o malou změnu polohy zrcátek promítající stupnice, kterou lze při odaretovaných vahách upravit korigačním šroubem na pravé zadní straně vah.
Pokud na misku vah vložíme nějaký předmět, je rovnováha porušena a k jejímu obnovení musíme ze závěsu vyzvednout odpovídající závaží. K tomu slouží ovládací mechanismum na levé dolní straně skříně vah. Skládá se ze tří soustředných šroubů. Pravým vnitřním ovládáme desítky gramů, středním jednotky a vnějším levým pak desetiny gramů. Hodnoty sejmutých závaží se ukazují v okénku stupnice. Pokud jsou sejmuta závaží zhruba odpovídající skutečné hmotnosti váženého předmětu, ukáže se hodnota setin a tisícin na světelné stupnici po odaretování vah. Desetitisíciny odečteme na základě nonia, tzn. že pravý malý šroub vedle stupnice desetitisícin nastavíme tak, aby ryska na světelné stupnici byla vyrovnána s nejbližší možnou hodnotou tisícin. Na stupnici desetitisícin pak odečteme danou hodnotu.
Pro veškeré vážení platí základní pravidlo: chemikálie (kapalné ani pevné) nesmí přijít do přímého styku s miskami vah! Veškerá manipulace s chemikáliemi (přidávání a ubírání) se musí provádět zásadně mimo váhy. Případné nečistoty na vahách je třeba okamžitě odstranit štětečkem.
ÚKOL č. 1: Určete průměrnou hmotnost a chybu měření váženého předmětu
POSTUP
Na analytických vahách postupně určete hmotnost deseti stejných předmětů (porcelánový kelímek, mikrozkumavka, kyveta) a určete aritmetickou průměrnou hmotnost jednoho předmětu a chybu měření. Vážení provádějte následovně:
1/ Stanovte, případně upravte, nulovou polohu vah. Aretační šroub má dvě polohy. Váhy jsou plně odaretovány až ve druhé spodní poloze!
2/ Vážený předmět zvažte na předvážkách na desetiny gramů.
3/ Vážený předmět vložte na misku zaaretovaných vah a nastavte závaží souhlasně s hodnotou získanou na předvážkách.
17

4/ Opatrně odaretujte. Pokud dojde k prudkému pohybu světelné stupnice a stupnice se dostane mimo rozsah, váhy zpátky zaaretujte a nastavte závaží o desetinu gramu vyšší či nižší.
5/ Toto provádějte tak dlouho, až se vám podaří světelnou stupnici nastavit v jejím rozsahu. Vyčkejte až do ustálení rovnováhy.
6/ Poté dolaďte šroubem na desetitisíciny gramu rysku souhlasně se stupnicí a odečtěte získanou hodnotu s přesností na čtyři desetinná místa.
Nezapomínejte vždy při odaretovaných vahách zavírat obě boční dvířka, aby nebyla rovnováha rušena okolním prouděním vzduchu.
7/ Váhy zaaretujte a vyjměte vážený předmět. Po skončeném vážení je třeba vrátit všechna závaží zpět a překontrolovat nulovou polohu vah.
VYHODNOCENÍ
Při měření se můžete dopustit nahodilých a systematických chyb. Zatímco systematické chyby mohou být zapříčiněny například nevhodným postupem při měření, špatnou kalibrací apod. a nelze je odstranit opakovaným měřením za stále stejných podmínek, chyby nahodilé můžete výpočtem konečné hodnoty z dostatečně velkého počtu měření z větší části eliminovat. Opakovaným měřením se vliv nahodilých chyb zmenší.
Měřenou veličinu označíte A, při n počtu měření naměříte postupně hodnoty A1 až An. Nejlepším odhadem veličiny A je pak průměr nalezených hodnot Â.
n
Ai Â
n
1i∑==
Čím je n větší, tím se hodnota  více blíží správné hodnotě A. Jestliže je odchylka každého měření od této střední hodnoty označená xi: Â−= ii Ax
pak je střední chybu výsledku (aritmetického průměru) definovaná takto:
11
2
−=∑=
n
xn
ii
δ
a výsledek se píše ve tvaru: δ±= ÂA
Výsledky měření zapište do tabulky a určete aritmetickou průměrnou hmotnost jednoho předmětu a střední chybu výsledku.
MĚŘENÍ OBJEMŮ KAPALIN
K odměřování objemů kapalin slouží odměrné válce, pipety, byrety, pyknometry a odměrné baňky.
Odměrné válce (obr. 6-1) se používají jen k přibližnému odměřování kapalin.
Na přesnější měření objemů se používají pipety (buď pouze pro určitý objem, obr. 6-2, nebo dělené obr. 6-3,4) a byrety u nichž je možno kohoutkem nebo pomocí tlačky regulovat vytékání kapaliny (obr. 6-5). Všechny tyto nádoby jsou kalibrovány na vylití, tzn., že z nich vyteče
18

přesně vyznačený objem. Při plnění pipet je třeba vždy dbát toho, aby ústí pipety bylo stále ponořeno pod hladinou kapaliny. Při jeho vynoření nad hladinu dochází k nasátí vzduchu do pipety, což může zapříčinit i vniknutí kapaliny do úst. Po nasátí kapaliny nad rysku označující požadovaný objem uzavřeme horní konec pipety ukazovákem – ne palcem. A opatrným uvolňováním prstu po kapkách vypouštíme kapalinu. Při odečítání je nutno mít oko ve stejné úrovni se značkou. Odečítáme vždy spodní okraj menisku.
1 2 3 4 5 6 7
Obrázek 6: Pomůcky k měření objemů. 1. kalibrovaný odměrný válec, 2. odměrné baňky, 3. pipeta, 4. pipeta dělená, 5. byrety, 6. automatická pipeta, 7. automatická pipeta multikanálová.
Jedovaté kapaliny a koncentrované kyseliny a zásady nenasáváme nikdy do pipety ústy, ale používáme speciální násadky či gumové balónky. Jelikož je pipeta kalibrována na vylití, nikdy se nevyfukuje, ale její obsah se nechá pouze vytéct a její špička se otře o dno nebo o stěnu nádobky, do které kapalinu odměřujeme.
V současné době se v biochemické či molekulárně biologické laboratoři se skleněnými pipetami téměř nesetkáváme, plně je nahradily pipety automatické (obr. 2-6,7) umožňující přesnější a pohodlnější odměření daného objemu. Jsou vhodné i pro pipetování mikrolitrových objemů. Automatické pipety jsou vyráběny pro různé rozsahy objemů (5-1 mL, 1000-200 µL, 200-20 µL, 20-2 µL a 2-0,2 µL). Objem se nastavuje otočením šroubu na stupnici. Nasávání a vypouštění kapaliny přes nasaditelnou špičku je ovládáno pístem, který má tři polohy – klidovou, pro nasávání a pro vypouštění (obr. 7).
klidová poloha poloha pro nasávání poloha pro vypouštění
Obrázek 7: Polohy automatické pipety při nasávání a vypouštění kapaliny.
19

Byrety, sloužící k regulovanému odběru kapalin při titracích (obr. 6-5), jsou skleněné trubice opatřené dělícími značkami. Před výtokem je umístěn kohoutek nebo pružná tlačka s hadičkou. Některé byrety mají pro lepší odečítání objemu zadní stěnu z bílého skla s modrým pruhem ve středu.
Odměrné baňky (obr. 6-2), stejně jako pyknometry (nádobky pro stanovování hustoty), jsou kalibrovány na dolití – to znamená, že při jejich naplnění obsahují právě požadovaný objem. Hrdlo odměrných baněk je poměrně úzké, po celém obvodu opatřené ryskou. I zde je třeba plnit tak, aby se spodní okraj menisku dotýkal rysky a při plnění mít oko v úrovní rysky. Podobně jako odměrné válce i odměrné baňky se vyrábějí v objemech od 5 mL do 2 L. Odměrné baňky se používají k přípravě roztoků o přesné koncentraci. Vlastní směšovaní složek roztoku provádíme při ne zcela zaplněné baňce a teprve po úplné homogenizaci a vyrovnání teplot (teplota na kterou je baňka kalibrována, je uváděna na plášti baňky - většinou 20°C) opatrně doplňujeme rozpouštědlem po rysku.
ÚKOL č. 2: Práce s automatickou pipetou
Seznamte se se sadou automatických pipet a vyzkoušejte si princip, na kterém fungují.
Připravte tři suché 25 mL kádinky. Kádinku předem zvažte na analytických vahách, a poté do ní napipetujte pipetou z rozsahem 1-5 mL desetkrát po jednom mililitru destilované vody a opět zvažte. Stejně postupujte s pipetou o rozsahu 1000-100 µL. Pipetujte 10x po 200 µL a 10x po 400 µL. Do třetí zvážené kádinky, pak pipetujte 10x po 100µL a 10x po 20µL pipetou s rozsahem 200-20 µL.
Uvědomte si, jakou hustotu má destilovaná voda, a vypočítejte, jaké hmotnosti by měly mít kádinky s vodou. Pokud jste nedosáhli požadované hmotnosti, zopakujte pipetování znovu po konzultaci s vedoucím cvičení. PŘÍPRAVA PUFRŮ A MĚŘENÍ pH
Výběr vhodného pufru se řídí především požadovanou hodnotou pH, iontovou silou, možnými
interakcemi s ostatními komponentami reakční směsi apod. Má-li mít pufr dostatečnou pufrační kapacitu, musíme vybrat takovou slabou kyselinu nebo zásadu, jejíž pKa se neodlišuje od požadované hodnoty pH o více než 1. Pufrační kapacita závisí také na celkové koncentraci (molaritě) pufru. Očekáváme-li například, že pH roztoku v průběhu reakce bude klesat (např. při studiu lipasové reakce, kdy jsou do roztoku uvolňovány ionty H+ disociací vznikajících mastných kyselin), pak volíme pufr o vyšší koncentraci a s pKa pod hodnotou pH, v níž začínáme pokus.
Složení vhodného pufru lze nalézt v laboratorních příručkách. Podle tabulek smícháme příslušné (obvykle dva) roztoky pro dosažení nominální hodnoty pH. Vždy je nutné skutečnou hodnotu pH měřením ověřit a případně ji upravit roztokem kyseliny nebo zásady. Jednodušší je připravit pufr prostou úpravou pH roztoku zvolené soli, kyseliny nebo báze. Pro jeho požadovanou hodnotu vybereme vhodnou slabou kyselinu nebo zásadu (ev. je v návodu pro práci s daným materiálem určena) a vypočteme navážku tak, abychom získali potřebný objem pufru o zvolené koncentraci. Navážené množství rozpustíme v menším objemu vody (asi 80 % konečného objemu). Pak nastavíme požadované pH roztokem silné (v některých případech i slabé) kyseliny či zásady a nakonec doplníme vodou na vypočtený objem. Pouze při užití pufrů o více složkách (pokrývajících široké rozmezí pH) nelze takto postupovat a musíme je namíchat podle tabulky, zkontrolovat a eventuálně upravit pH.
Většina pufrů podléhá mikrobiální kontaminaci (nejen organické kyseliny, ale i např. fosfát zplesniví při 0-4 oC během týdne). Pro některé účely je lze konzervovat (azid, toluen, thymol),
20

většinou jsou ale uchovávány v chladničce bez konzervace. Takto skladujeme jen menší množství připraveného pufru, které brzy spotřebujeme (maximálně do týdne). Připravíme-li si větší množství pufru do zásoby, uchováváme ho zmrazený v plastové láhvi. Před použitím je nutno rozmrazit celé množství pufru a roztok zamíchat. Po odebrání potřebného objemu je možné zbytek opět zmrazit. Popřípadě můžeme pufry autoklávovat. Při manipulaci s takto ošetřenými roztoky musíme dodržovat sterilní podmínky a pufr zpětně nekontaminovat, např. nesterilní špičkou automatické pipety.
Hodnotu pH roztoku ovlivňuje jeho ředění, zejména v extrémních hodnotách stupnice (pod 3 a nad 11), v rozmezí fyziologických hodnot již méně. Proto se raději vyhneme přípravě koncentrovanějších zásobních roztoků, z nichž bychom pak ředěním připravovali pracovní roztok. Také teplota ovlivňuje pH pufru změnou pKa slabé kyseliny nebo báze. Tabelované hodnoty pH jsou obvykle vztaženy na 18, 20 nebo 25 oC. V biochemii často pracujeme za chladu, kdy se pH může lišit od hodnoty nastavené při přípravě pufru za teploty místnosti. Např. měření pH závislosti enzymové reakce by mohlo být více či méně zkresleno, pokud se podstatně liší teplota experimentu od teploty, při které byl pufr připraven. V případě nutnosti lze upravit hodnotu pH za příslušné teploty.
Běžné pufry užívané v biochemických laboratorních postupech pokrývají neutrální až mírně alkalickou oblast o pH 6,0 – 9,0. Některé operace se provádějí v prostředích více kyselých či bazických. Nejběžnější kyseliny a báze, užívané k přípravě pufrů v biochemii, jsou v přehledu uvedeny v tabulce 1. Vedle požadované hodnoty pH se výběr složek řídí také jejich možnými specifickými vlivy na reakční směs. Někdy může dojít k vysrážení některých komponent směsi pufrem (K+-chloristanem, soli železa a Ca2+-fosfátem, komplexy borátů se sacharidy apod.), k interakci složek pufru s bílkovinami (Cl- iont s albuminem), k reakci s analytickými činidly a k inhibici enzymů (v některých případech barbituráty). Naopak někdy mohou být složky pufru využity jako substráty (citrát, acetát) aj. Pro biochemické účely se používají vedle běžných anorganických a organických látek i speciální kyseliny a báze, vhodné pro jejich inertnost k dalším složkám reakčních směsí (viz vysvětlivky pod tabulkou 1).
Koncentrace H+ iontů v roztoku se stanovuje spektrálními či elektrochemickými metodami. První z nich využívají toho, že změna pH vyvolá v některých organických sloučeninách (acidobazických indikátorech) změnu struktury lišící se spektrálními vlastnostmi (absorpce či fluorescence). Ve druhém případě se využívá změny elektrochemického potenciálu vlivem pH.
Nejjednodušší spektrální metody měření pH jsou vizuální metody, kdy se sleduje změna zbarvení indikátoru při změně pH. K této změně dochází u jednotlivých indikátorů v určité oblasti pH (nalezneme v chemických laboratorních tabulkách). Pro stanovení hodnoty pH musíme vybrat indikátor, u kterého dochází k barevné změně v přesně definovaném intervalu pH. Jsou vhodné pro indikaci dosažení požadované hodnoty pH, pro indikaci bodu ekvivalence při acidobazických titracích, kontrolu udržování pH apod. Používají se ve formě vodného či ethanolického roztoku (0,04 - 0,10 %), který se přikápne ke sledovanému vzorku. Pro universální použití slouží směsi indikátorů pokrývající svými barevnými přechody celou škálu pH nebo její část, která nás zajímá. Užívají se ve formě papírků napuštěných příslušnou směsí indikátorů. Souprava je doplněna barevnou stupnicí, s kterou porovnáváme zbarvení papírku po namočení do vzorku a ustálení zbarvení.
Barevný přechod indikátoru můžeme přesněji sledovat pomocí spektrofotometrických přístrojů. To umožňuje též kvantifikovat barevný přechod při změně pH (stanovení pKa). Tento způsob však není běžně užíván, je omezen na speciální případy (např. měření pH uvnitř buněk a organel nebo v nevodných rozpouštědlech). Při použití fluorescenčních indikátorů jsme zcela odkázáni na měření pomocí přístrojů – fluorimetrů. Tento způsob nemá visuální alternativu.
21
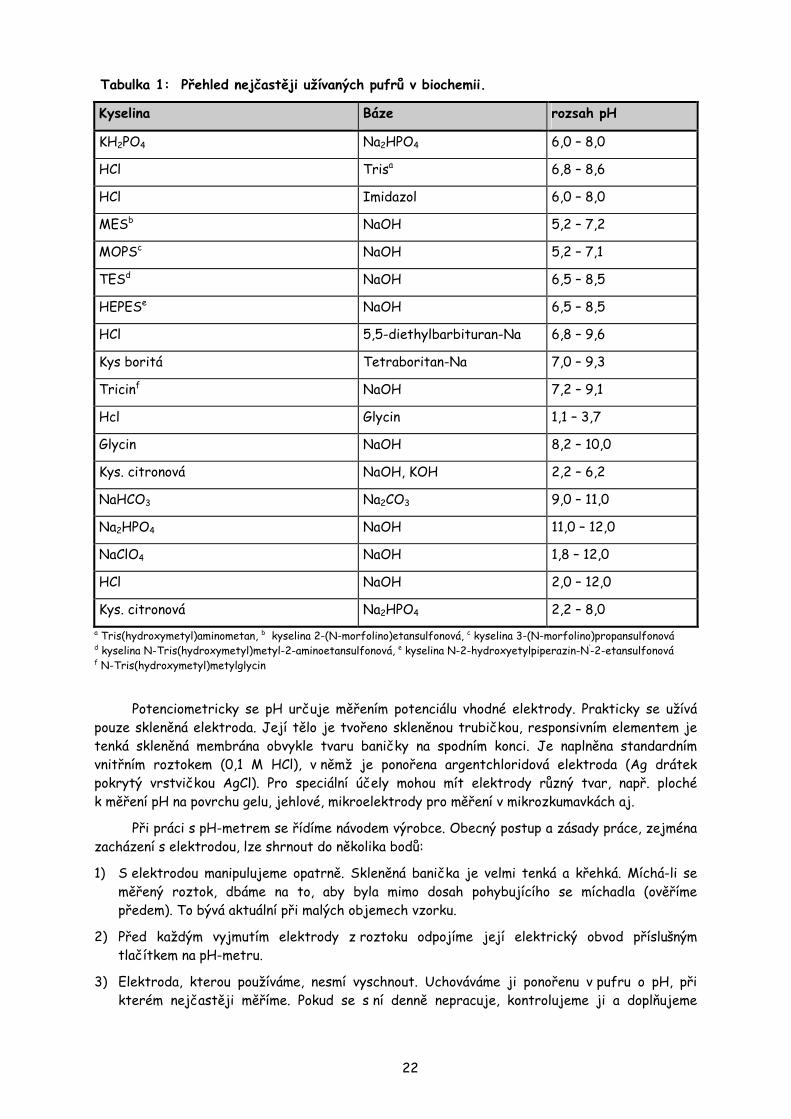
Tabulka 1: Přehled nejčastěji užívaných pufrů v biochemii.
Kyselina Báze rozsah pH
KH2PO4 Na2HPO4 6,0 – 8,0
HCl Trisa 6,8 – 8,6
HCl Imidazol 6,0 – 8,0
MESb NaOH 5,2 – 7,2
MOPSc NaOH 5,2 – 7,1
TESd NaOH 6,5 – 8,5
HEPESe NaOH 6,5 – 8,5
HCl 5,5-diethylbarbituran-Na 6,8 – 9,6
Kys boritá Tetraboritan-Na 7,0 – 9,3
Tricinf NaOH 7,2 – 9,1
Hcl Glycin 1,1 – 3,7
Glycin NaOH 8,2 – 10,0
Kys. citronová NaOH, KOH 2,2 – 6,2
NaHCO3 Na2CO3 9,0 – 11,0
Na2HPO4 NaOH 11,0 – 12,0
NaClO4 NaOH 1,8 – 12,0
HCl NaOH 2,0 – 12,0
Kys. citronová Na2HPO4 2,2 – 8,0 a Tris(hydroxymetyl)aminometan, b kyselina 2-(N-morfolino)etansulfonová, c kyselina 3-(N-morfolino)propansulfonová d kyselina N-Tris(hydroxymetyl)metyl-2-aminoetansulfonová, e kyselina N-2-hydroxyetylpiperazin-N’-2-etansulfonová f N-Tris(hydroxymetyl)metylglycin
Potenciometricky se pH určuje měřením potenciálu vhodné elektrody. Prakticky se užívá pouze skleněná elektroda. Její tělo je tvořeno skleněnou trubičkou, responsivním elementem je tenká skleněná membrána obvykle tvaru baničky na spodním konci. Je naplněna standardním vnitřním roztokem (0,1 M HCl), v němž je ponořena argentchloridová elektroda (Ag drátek pokrytý vrstvičkou AgCl). Pro speciální účely mohou mít elektrody různý tvar, např. ploché k měření pH na povrchu gelu, jehlové, mikroelektrody pro měření v mikrozkumavkách aj.
Při práci s pH-metrem se řídíme návodem výrobce. Obecný postup a zásady práce, zejména zacházení s elektrodou, lze shrnout do několika bodů:
1) S elektrodou manipulujeme opatrně. Skleněná banička je velmi tenká a křehká. Míchá-li se měřený roztok, dbáme na to, aby byla mimo dosah pohybujícího se míchadla (ověříme předem). To bývá aktuální při malých objemech vzorku.
2) Před každým vyjmutím elektrody z roztoku odpojíme její elektrický obvod příslušným tlačítkem na pH-metru.
3) Elektroda, kterou používáme, nesmí vyschnout. Uchováváme ji ponořenu v pufru o pH, při kterém nejčastěji měříme. Pokud se s ní denně nepracuje, kontrolujeme ji a doplňujeme
22

odpařenou vodu. Nemá-li se s ní pracovat delší dobu (např. přes prázdniny), je lépe ji omýt destilovanou vodou a po uschnutí vysušit a bezpečně uložit.
4) Suchou elektrodu (novou nebo dlouhodobě uloženou) je nutno před užitím připravit dle návodu výrobce. Obvykle se máčí několik hodin v 0,1 M HCl.
5) Referenční kalomelová elektroda může být samostatná nebo kombinovaná se skleněnou. Její těleso tvaru skleněné trubičky je vyplněno vnitřním roztokem (KCl o různé koncentraci, obvykle nasycený roztok) a vodivé spojení s vnějším měřeným roztokem zajišťuje (nejde-li o otevřenou elektrodu) průlinčitá přepážka (frita). Ta je u samostatné elektrody na spodním konci. U kombinované elektrody tvoří referenční elektroda plášť kolem skleněné a frita je umístěna na boku tohoto pláště nad baničkou skleněné elektrody. V horní části tělesa kalomelové elektrody je uzavřený otvůrek, jímž v případě potřeby doplňujeme vnitřní roztok tak, aby bylo zajištěno vodivé spojení s vývodem na konektor. V případě, že máme nasycenou kalomelovou elektrodu, se tímto otvůrkem doplňuje pevný KCl tak, aby na dně kalomelové elektrody vždy bylo možno zahlédnout několik krystalů.
6) Před měřením je nutno přístroj s elektrodou kalibrovat. Elektrodu vyjmeme z roztoku, v kterém je uchovávána (nezapomeneme rozpojit obvod), opláchneme destilovanou vodou, odsajeme přebytek vody (filtračním papírem nebo buničitou vatou) a ponoříme do standardního pufru o pH nejbližším tomu, který bude mít měřený roztok (obvykle bývá k disposici sada standardních pufrů o pH 4, 7 a 9). Současně ponoříme i referentní elektrodu. Používáme-li kombinovanou elektrodu, zkontrolujeme, zda je boční vývod referentní elektrody zcela ponořen pod hladinu. Nastavíme teplotní korekci na teplotu pufru a kalibračním knoflíkem nastavíme na stupnici nebo digitálním výstupu hodnotu pH standardního pufru. Elektrodu pak vyjmeme a po opláchnutí ji ponoříme do měřeného vzorku. Jeho pH nám ukáže výchylka či digitální výstup pH-metru. Kalibraci je nutno provádět vždy po zapnutí přístroje nebo měníme-li pH měřených vzorků o více než 1. Též při dlouhodobých měřeních ji občas zkontrolujeme.
7) Některé přístroje umožňují nebo dokonce vyžadují kalibraci na dva pufry, v tomto případě můžeme pracovat v širším rozmezí pH, aniž bychom vždy znovu museli přístroj kalibrovat. Zvolíme si vhodné pufry ohraničující oblast, v které hodláme pracovat. Na jeden z nich nastavíme výchozí hodnotu. Po ponoření elektrod do druhého pufru nastavíme jeho hodnotu jiným knoflíkem, kterým nastavujeme směrnici závislosti potenciálu na pH. Ta by měla být - 59 mV.pH-1, její podstatný pokles indikuje malou citlivost elektrody a je třeba zvážit její výměnu, event. úpravu.
ÚKOL č. 3: Připravte 1/4 litru 0,1M Tris/acetátového pufru o pH 7,6 a 1/4 litru 0,02M K-fosfátového pufru, pH 7,0
POSTUP
1/ Vypočtěte hmotnost Tris báze - tris(hydroxymetyl)aminomethan (Mr = 121,14), potřebné na přípravu 250 mL pufru o koncentraci 0,1 mol.L-1.
2/ Odvažte toto množství na předvážkách na jedno desetinné místo do 500 mL kádinky.
3/ Do kádinky přilijte asi 400 mL destilované vody, vložte elektromagnetické míchadlo a na elektromagnetické míchačce míchejte až do úplné homogenizace roztoku.
4/ Podle návodu nakalibrujte pH metr a poté upravte pH roztoku na hodnotu 7,6 pomocí koncentrované kyseliny octové. Přídavky kyseliny provádějte pomocí Pasteurovy pipety za stálého míchání roztoku.
23
5/ Až se hodnota pH stabilizuje (asi jednu minutu je konstantní) vyjměte elektrodu, řádně ji opláchněte a vraťte ji do ochranného obalu s roztokem.
6/ Pufr z kádinky přelijte kvantitativně pomocí nálevky do 500 mL odměrné baňky a opatrně doplňte objem po rysku střičkou s destilovanou vodou.
7/ Hotový pufr přelijte do zásobní láhve.
Stejně postupujte při přípravě 250 mL 0,02 M K-fosfátového pufru, navážky kyselého (KH2PO4,
Mr = 136,09) a bazického (K2HPO4, Mr = 174,18) fosforečnanu si vypočítejte podle následující tabulky:
Tabulka 2: Příprava K-fosfátového pufru. Na 0,2 M pufr je třeba smíchat: x mL 0,2 M bazického roztoku fosforečnanu a y mL 0,2 M kyselého roztoku fosforečnanu.
pH x 0.2 M K2HPO4 (mL) y 0.2 M KH2PO4 (mL)
6,0 12,3 87,7
6,5 31,5 68,5
7,0 61,0 39,0
7,5 84,0 16,0
8,0 94,7 5,3
Hodnotu pH pufru zkontrolujte na pH metru a případnou odchylku od pH 7,0 upravte kyselinou fosforečnou nebo hydroxidem draselným.
PŘÍPRAVA ROZTOKŮ
Základním pravidlem pro jakoukoli přípravu roztoků, vzorků pro analýzu nebo pro účely
preparace nebo čištění sloučenin je, že se rozpouštěná látka přidává do vody a nikdy naopak. Při přípravě odměrných roztoků přenášíme kvantitativně navážku nebo odměřené množství kapaliny do vody, která zaujímá asi 1/3 – 1/2 objemu baňky. Kvantitativní přenesení znamená, že navážku krystalické látky sklepneme do malé nálevky zasunuté do hrdla odměrné baňky, spláchneme větším množstvím destilované vody ze střičky, vodou ze střičky dále do nálevky vydatně opláchneme navažovací lodičku a nakonec nálevku vypláchneme vodou ze střičky po celém vnitřním povrchu. Rozpouštění můžeme urychlovat pouze mícháním krouživým pohybem celé odměrné baňky, nikdy ne tyčinkou nebo míchadlem. Teprve po úplném rozpuštění navážky a vyrovnání teploty baňky s okolím můžeme baňku doplnit po rysku destilovanou vodou (poslední kapky přidáváme pipetou). Pokud je látka, jejíž roztok připravujeme, kapalná, funkci navažovací lodičky může převzít malý odměrný válec, který se pak rovněž do odměrné baňky vyplachuje. Pokud kapalnou složku do vody v odměrné baňce pipetujeme, jsme problémů s kvantitativním přenesením vzorku ušetřeni. Po doplnění odměrné baňky po rysku nezapomeneme obsah důkladně promíchat. Naplněnou baňku uzavřeme čistou, suchou, v současné době nejčastěji polyethylenovou zátkou a několikrát obrátíme dnem vzhůru a zpět. Zátku přitom stále v hrdle baňky přidržujeme palcem.
ÚKOL č. 4: Příprava koncentrační řady roztoku kyseliny askorbové pro kinetická měření
Při biochemických experimentech se často setkáte s potřebou připravit sestupnou koncentrační řadu roztoků o přesných koncentracích, jako třeba při měření kinetických parametrů různých enzymových systémů, kdy se měří enzymatická aktivita při různé koncentraci substrátu v reakční směsi. Jako příklad lze uvést měření Michaelisovy konstanty KM, což je konstanta udávající koncentraci substrátu za daných podmínek, při které enzymová reakce probíhá polovinou maximální rychlosti. Pro toto stanovení je nutno změřit aktivitu enzymu při
24
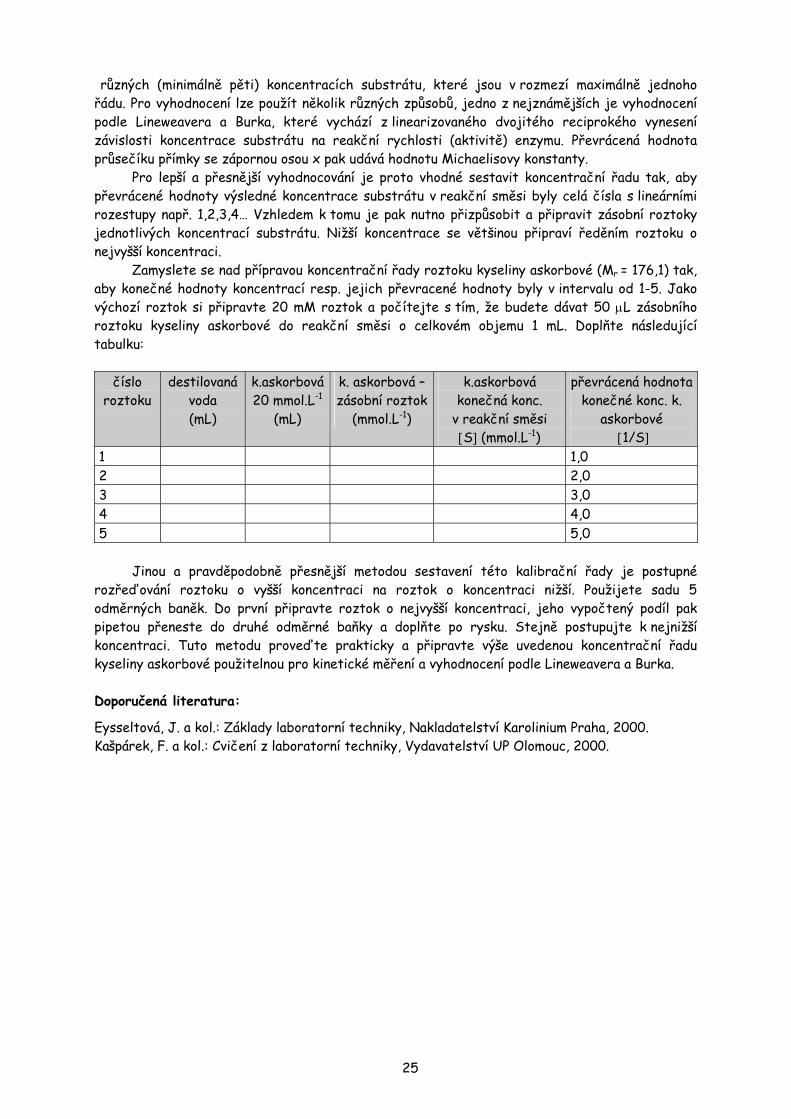
různých (minimálně pěti) koncentracích substrátu, které jsou v rozmezí maximálně jednoho řádu. Pro vyhodnocení lze použít několik různých způsobů, jedno z nejznámějších je vyhodnocení podle Lineweavera a Burka, které vychází z linearizovaného dvojitého reciprokého vynesení závislosti koncentrace substrátu na reakční rychlosti (aktivitě) enzymu. Převrácená hodnota průsečíku přímky se zápornou osou x pak udává hodnotu Michaelisovy konstanty.
Pro lepší a přesnější vyhodnocování je proto vhodné sestavit koncentrační řadu tak, aby převrácené hodnoty výsledné koncentrace substrátu v reakční směsi byly celá čísla s lineárními rozestupy např. 1,2,3,4… Vzhledem k tomu je pak nutno přizpůsobit a připravit zásobní roztoky jednotlivých koncentrací substrátu. Nižší koncentrace se většinou připraví ředěním roztoku o nejvyšší koncentraci.
Zamyslete se nad přípravou koncentrační řady roztoku kyseliny askorbové (Mr = 176,1) tak, aby konečné hodnoty koncentrací resp. jejich převracené hodnoty byly v intervalu od 1-5. Jako výchozí roztok si připravte 20 mM roztok a počítejte s tím, že budete dávat 50 µL zásobního roztoku kyseliny askorbové do reakční směsi o celkovém objemu 1 mL. Doplňte následující tabulku:
číslo
roztoku destilovaná
voda (mL)
k.askorbová 20 mmol.L-1
(mL)
k. askorbová – zásobní roztok
(mmol.L-1)
k.askorbová konečná konc.
v reakční směsi [S] (mmol.L-1)
převrácená hodnota konečné konc. k.
askorbové [1/S]
1 1,0 2 2,0 3 3,0 4 4,0 5 5,0
Jinou a pravděpodobně přesnější metodou sestavení této kalibrační řady je postupné
rozřeďování roztoku o vyšší koncentraci na roztok o koncentraci nižší. Použijete sadu 5 odměrných baněk. Do první připravte roztok o nejvyšší koncentraci, jeho vypočtený podíl pak pipetou přeneste do druhé odměrné baňky a doplňte po rysku. Stejně postupujte k nejnižší koncentraci. Tuto metodu proveďte prakticky a připravte výše uvedenou koncentrační řadu kyseliny askorbové použitelnou pro kinetické měření a vyhodnocení podle Lineweavera a Burka.
Doporučená literatura: Eysseltová, J. a kol.: Základy laboratorní techniky, Nakladatelství Karolinium Praha, 2000. Kašpárek, F. a kol.: Cvičení z laboratorní techniky, Vydavatelství UP Olomouc, 2000.
25

Laboratorní cvičení č. 3
ÚPRAVA BIOLOGICKÉHO MATERIÁLU, PRECIPITAČNÍ TECHNIKY, DIALÝZA, CENTRIFUGACE, ULTRAFILTRACE.
ÚPRAVA BIOLOGICKÉHO MATERIÁLU
K biochemickým studiím se používá nejrůznější biologický materiál, který příroda poskytuje. Vzhledem k jeho různorodosti je technika zpracování mnohdy velmi odlišná a řídí se účelem, k jakým studiím má být použit. Způsob zpracování materiálů bývá často odlišný podle toho, zda má být výchozí surovinou pro izolaci, detekci nějaké sloučeniny či skupiny látek nebo zda chceme studovat jejich metabolismus.
Má-li být izolována nějaká látka nebo skupina látek z biologického materiálu (např. vitamín, bílkovina, sacharid apod.) je důležité zvolit vhodnou výchozí surovinu. Pro izolaci se hodí nejlépe takový biologický materiál, v kterém je požadovaná látka v nejvyšší koncentraci, je dostupný a jeho cena přijatelná. Vybraný živočišný, rostlinný nebo mikrobiální materiál je třeba v prvním kroku zhomogenizovat. Rozrušení rostlinných a živočišných buněk nedělá obvykle potíže. Jinak však je tomu při izolacích z buněk mikroorganismů. Buněčná stěna mikroorganismů je velmi rezistentní a její narušení vyžaduje zvláštní postupy, viz níže.
1) Mletí Suchý materiál může být rozemlet v různých typech mlýnků. Semena se melou v kulových
mlýnech nebo mlýnech s otočnými noži. K mletí živočišných materiálů se velmi dobře hodí masový strojek. Tkáň se v něm řeže a drtí. Hrubost nebo jemnost lze regulovat vložením vhodné destičky, kterou se rozemletý materiál protlačuje. Všechny uvedené strojky a mlýnky většinou může nahradit běžný univerzální kuchyňský robot, který je vybaven mixerem, mlýnkem, masovým strojkem i struhadly.
2) Homogenizace
Na rozdíl od mletí nasucho je v biochemické práci chápána homogenizace jako důkladné rozmělnění materiálu za přídavku vody nebo vhodných pufru či fyziologických roztoků.
Pro homogenizaci malých množství je nejvhodnější roztírání materiálu ve třecí misce za přídavku mořského písku, skelného prachu nebo jiného abraziva. Tužší materiály se před homogenizací zmrazí kapalným dusíkem, který je učiní křehčími k rozmělnění. Homogenizaci lze rovněž provést ve výkonném mixeru.
K narušení buněčné stěny a membrány mikroorganismů, tzv. desintegraci, se používá ještě některých speciálních metod. Např. působení chemických činidel (detergenty), působení enzymů štěpích polysacharidy buněčných stěn (lysozym, celulasa), krátké působení ultrazvuku, opakované zmrazování a rozmrazování. Přehled nejužívanějších homogenizačních technik je uveden v tabulce 3.
PRECIPITAČNÍ METODY
V počátečních fázích izolací biopolymerů jsou většinou používány méně pracné metody fázové separace, jako jsou precipitace (frakcionace), dialýza či vsádková adsorpce. Tyto metody nemají zpravidla velké rozlišení, umožňují však snadné a rychlé zakoncentrování preparátu po základní extrakci biologického materiálu. Srážení se provádí pomocí neutrálních solí, změnou pH, organickými rozpouštědly či teplotou.
26
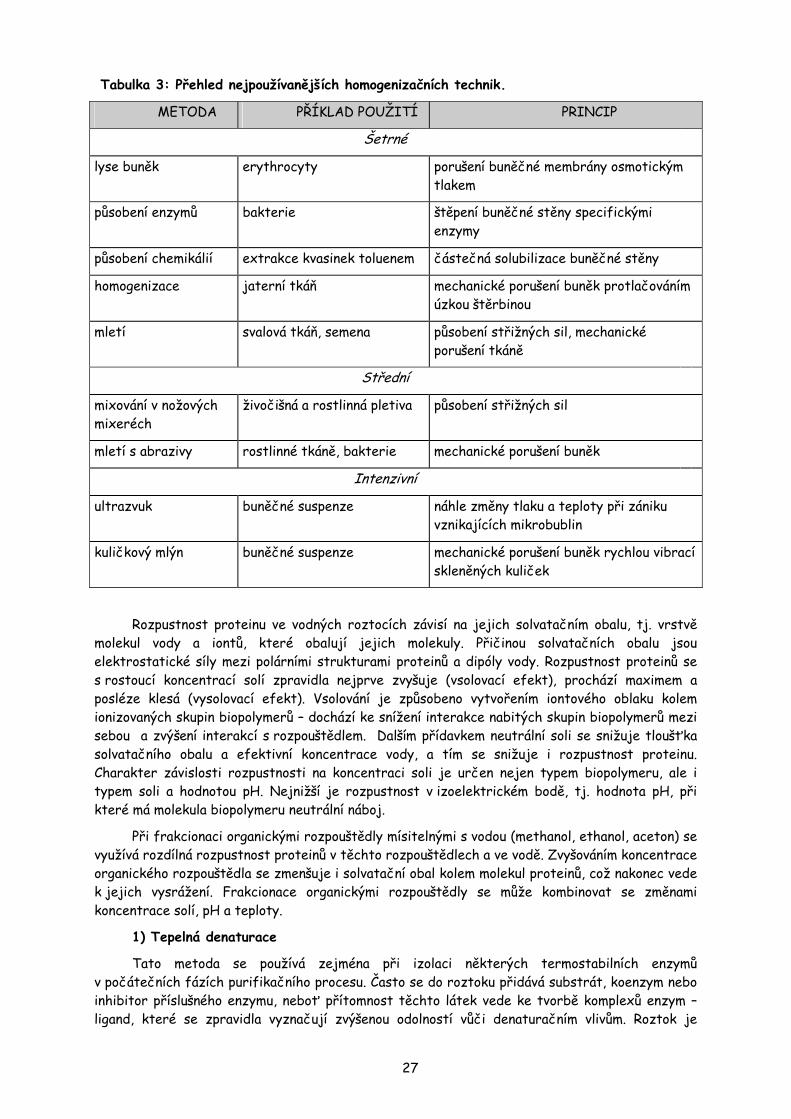
Tabulka 3: Přehled nejpoužívanějších homogenizačních technik.
METODA PŘÍKLAD POUŽITÍ PRINCIP
Šetrné
lyse buněk erythrocyty porušení buněčné membrány osmotickým tlakem
působení enzymů bakterie štěpení buněčné stěny specifickými enzymy
působení chemikálií extrakce kvasinek toluenem částečná solubilizace buněčné stěny
homogenizace jaterní tkáň mechanické porušení buněk protlačováním úzkou štěrbinou
mletí svalová tkáň, semena působení střižných sil, mechanické porušení tkáně
Střední
mixování v nožových mixeréch
živočišná a rostlinná pletiva působení střižných sil
mletí s abrazivy rostlinné tkáně, bakterie mechanické porušení buněk
Intenzivní
ultrazvuk buněčné suspenze náhle změny tlaku a teploty při zániku vznikajících mikrobublin
kuličkový mlýn buněčné suspenze mechanické porušení buněk rychlou vibrací skleněných kuliček
Rozpustnost proteinu ve vodných roztocích závisí na jejich solvatačním obalu, tj. vrstvě molekul vody a iontů, které obalují jejich molekuly. Přičinou solvatačních obalu jsou elektrostatické síly mezi polárními strukturami proteinů a dipóly vody. Rozpustnost proteinů se s rostoucí koncentrací solí zpravidla nejprve zvyšuje (vsolovací efekt), prochází maximem a posléze klesá (vysolovací efekt). Vsolování je způsobeno vytvořením iontového oblaku kolem ionizovaných skupin biopolymerů – dochází ke snížení interakce nabitých skupin biopolymerů mezi sebou a zvýšení interakcí s rozpouštědlem. Dalším přídavkem neutrální soli se snižuje tloušťka solvatačního obalu a efektivní koncentrace vody, a tím se snižuje i rozpustnost proteinu. Charakter závislosti rozpustnosti na koncentraci soli je určen nejen typem biopolymeru, ale i typem soli a hodnotou pH. Nejnižší je rozpustnost v izoelektrickém bodě, tj. hodnota pH, při které má molekula biopolymeru neutrální náboj.
Při frakcionaci organickými rozpouštědly mísitelnými s vodou (methanol, ethanol, aceton) se využívá rozdílná rozpustnost proteinů v těchto rozpouštědlech a ve vodě. Zvyšováním koncentrace organického rozpouštědla se zmenšuje i solvatační obal kolem molekul proteinů, což nakonec vede k jejich vysrážení. Frakcionace organickými rozpouštědly se může kombinovat se změnami koncentrace solí, pH a teploty.
1) Tepelná denaturace
Tato metoda se používá zejména při izolaci některých termostabilních enzymů v počátečních fázích purifikačního procesu. Často se do roztoku přidává substrát, koenzym nebo inhibitor příslušného enzymu, neboť přítomnost těchto látek vede ke tvorbě komplexů enzym – ligand, které se zpravidla vyznačují zvýšenou odolností vůči denaturačním vlivům. Roztok je
27
zahřát po jistou dobu na určitou teplotu, pak je rychle ochlazen v ledové lázni a koagulované balastní biopolymery, zejména termolabilní proteiny, se odstraní centrifugací nebo filtrací. Jako příklad můžeme uvést přípravu kvasničné alkoholdehydrogenasy, kdy se extrakt v prostředí pyrofosfátového pufru zahřeje po dobu 15 minut na teplotu 55°C na vodní lázní a balastní proteiny se odstraní centrifugací.
2) Vysolování neutrálními solemi
Precipitovatelnost určitého proteinu z vodného roztoku je závislá na druhu použité soli, iontové síle, hodnotě pH, teplotě a koncentraci proteinů. Při vysolovacím efektu se uplatňuje daleko více povaha aniontu než kationtu. Dvojmocné a vícemocné anionty precipitují proteiny většinou mnohem účinněji než anionty jednomocné. Nejčastěji používanou solí pro vysolování biopolymerů je síran amonný. Jeho výhodou je značná rozpustnost (např. ve srovnání s Na2SO4), která je jen nepatrně závislá na teplotě, a malý denaturační vliv. Doporučuje se používat dostatečně čistý síran amonný bez obsahu kovových iontů, které by mohly interagovat s proteiny a katalyzovat některé oxidační reakce. Jiné soli se používají jen ve speciálních případech, např. síran hořečnatý k izolaci γ-globulinové frakce ze séra nebo chlorid sodný k precipitaci fibrinogenu. Díky své velké kapacitě a tlumivé schopnosti by byly fosforečnany ideálními srážedly, ale pro svou špatnou rozpustnost se v praxi používají jen velmi omezeně. Často používanou solí pro precipitaci některých enzymů je i chlorid manganatý.
Tabulka 4: Tabulka uvádí množství pevného síranu amonného, které je třeba přidat k jednomu litru roztoku, aby se dosáhlo žádané změny v procentech saturace roztoku síranem amonným.
Výsledná koncentrace síranu amonného (% saturace) 10 15 20 25 30 33 35 40 45 50 55 60 65 70 75 80 85 90 95 100
0 56 84 114 144 176 196 209 243 277 313 351 390 430 472 516 561 610 662 713 767 10 28 57 86 118 137 150 183 216 251 288 326 365 406 449 494 540 592 640 694 15 28 57 88 107 120 153 185 220 256 294 333 373 415 459 506 556 605 657 20 29 59 78 91 123 155 189 225 262 300 340 382 424 471 520 569 619 25 30 49 61 93 125 158 193 230 267 307 348 390 436 485 533 583 30 19 30 62 94 127 162 198 235 273 314 356 401 449 496 546 33 12 43 74 107 142 177 214 252 292 333 378 426 472 522 35 31 63 94 129 164 200 238 278 319 364 411 457 506 40 31 63 97 132 168 205 245 285 328 375 420 469 45 32 65 99 134 171 210 250 293 339 383 431 50 33 66 101 137 176 214 256 302 345 392 55 33 67 103 141 179 220 264 307 353 60 34 69 105 143 183 227 269 314 65 34 70 107 147 190 232 275 70 35 72 111 153 194 237 75 36 74 115 155 198 80 38 77 117 157 85 39 77 118 90 38 77 95 39
Výchozí koncentrace síranu amonného (% saturace)
Vlastní precipitace se provádí tak, že se do roztoku biopolymerů sůl zpravidla přidává po malých dávkách v pevném stavu nebo jako nasycený vodný roztok za stalého míchání, aby nedošlo k lokálnímu přesycení. Vysolování probíhá při konstantní teplotě, chlazení není vždy nezbytné. Optimální koncentrační rozsah pro frakcionaci daného proteinu je nutno najít empiricky. Množství síranu amonného, potřebného k dosažení určitého stupně nasycení, lze snadno zjistit z tabulky 4. Po přidání takového množství síranu amonného, které odpovídá 30% množství potřebného ke vzniku nasyceného roztoku této soli (v praxi označováno jako 30% nasycení), se
28

srážejí nukleové kyseliny a poměrně málo proteinů. Mezi 30 – 75% nasycení pak precipituje většina proteinů. V tomto rozmezí je třeba nasycení zvyšovat po poměrně malých dávkách. Po přidání příslušného množství soli se roztok biopolymeru míchá ještě asi 20 minut a precipitát se oddělí centrifugací. Pokud je daný biopolymer obsažen v precipitátu, lze po rozpuštění sraženiny získat jeho koncentrovaný roztok. Síran amonný se z roztoku oddělí většinou dialýzou, gelovou chromatografií nebo ultrafiltrací.
3) Srážení biopolymerů ostatními látkami
Poměrně starou metodou je precipitace biopolymerů organickými rozpouštědly. Tyto látky snižují dialektrickou konstantu prostředí, tím přispívají ke snížení rozpustnosti a k vysrážení biopolymeru (zvyšují se elektrostatické interakce mezi nabitými skupinami biopolymeru a molekuly se pak snáze shlukují do precipitujících agregátů). Při izolaci nukleových kyselin se používá srážení koncentrovaným ethanolem (pro kvantitavní odstranění nukleových kyselin z roztoku se často používají ribonukleasy a deoxyribonukleasy). Při separaci proteinů se organická rozpouštědla využívají málo, je zde nebezpečí denaturace, které se snižuje udržováním roztoku při teplotě 0°C. Ethanolová precipitace má výhodu v tom, že odpadá potřeba dialýzy. Kromě ethanolu se především pro frakcionaci plazmových (sérových) proteinů používá methanol, aceton a ether. Při separaci citlivějších proteinů jsou organická rozpouštědla nahrazována některými kyselinami, jako je kyselina kaprylová, rivanol (2-ethoxy-6,9-diaminoakridinlaktát) a nebo organickými polymery, např. polyethylenglykolem. Ke zvýšení specifičnosti srážení je možné do roztoku přidat nějaké specifické ligandy bílkovin, např. kofaktory nebo zinečnaté ionty. V poslední době se také začal pro některé kyselé proteiny používat bazický protamin sulfát, nízkomolekulární protein izolovaný z rybího spermatu. Princip kyselé či bazické frakcionace spočívá v pomalém okyselení či zalkalizování roztoku biopolymerů na určitou hodnotu pH pomocí silné kyseliny či zásady, na které se ponechá po určitou dobu (1 hod) a po odstranění vysrážených balastů vrátí na původní hodnotu.
Pokud nám jde o kvantitativní oddělení biopolymerů z roztoku, tzn. deproteinizaci, použijeme drastičtějších metod srážení. Lze využít např. srážecí schopnost iontů těžkých kovů (Hg2+, Pb2+, Cd2+) nebo složitějších aniontů, které tvoří s proteiny těžko rozpustné komplexy (kyseliny trichloroctová, pikrová, chloristá, fosfowolframová atd.). Tyto metody používáme tehdy když se nezajímáme o biopolymery, ale naopak o nízkomolekulární složky biologického materiálu.
CENTRIFUGACE
Centrifugace čili odstřeďování patří k nejběžnějším operacím v biochemické laboratoři. Dovoluje oddělit složky suspenze nebo emulze na základě rozdílných hustot. Bývá často rychlejší a pohodlnější než filtrace a v některých případech vede i k lepšímu oddělení pevné a kapalné fáze. Kromě nahrazení nebo doplnění filtrace, zvláště je-li suspendovaná látka velmi jemná, špatně filtrovatelná a filtrace zdlouhavá, má odstřeďování v biochemii i jiný význam. Slouží k některým speciálním preparativním a analytickým účelům (např. izolace mitochondrií diferenční centrifugací). Dále je to příprava buněčných struktur gradientovou centrifugací a z analytických aplikací pak především stanovení relativních molekulových hmotností sloučenin a stanovení čistoty izolovaných preparátů.
Při odstřeďování působí na sedimentující částice odstředivá síla (P), kterou lze vyjádřit vztahem:
P = m.r.ω2
kde m je hmotnost částice, r je poloměr otáčení a ω je úhlová rychlost. Pro praktické výpočty se však zavádí veličina relativní odstředivé zrychlení R, které udává, kolikrát je toto zrychlení větší než zemské gravitační zrychlení g a je dána vztahem:
R = 1,118.r.N2.10-5
29

kde N je počet otáček za minutu a r je poloměr otáčení v cm. Je nutno ji uvádět jako hlavní charakteristiku centrifugace. Ze vzorce je zřejmé, že pouhý údaj počtu otáček za minutu necharakterizuje proces centrifugace.
Aby se nemuselo v každém případě vypočítávat relativní odstředivé zrychlení, výrobci často dodávají k odstředivkám diagramy, z nichž se hodnota R snadno odvodí. Běžně se relativní odstředivé zrychlení odečítá také z univerzálního nomogramu, viz obrázek 8.
Existuje celá řada typu centrifug, které se liší velikostí (od malých stolních až po velkoobjemové průmyslové odstředivky), dosahovaným zrychlením a tvarem rotorů (výkyvné a úhlové). Ve výkyvných rotorech jsou kyvety uloženy v pouzdrech, která jsou volně zavěšena v čepech vlastního rotoru a kyvety jsou během odstřeďování ve vodorovné poloze. Naproti tomu v úhlových rotorech jsou kyvety fixovány v určitém úhlu (45 - 50°) k ose otáčení.
Zvláštní kategorii tvoří ultracentrifugy, na nichž lze dosáhnout relativního odstředivého zrychlení 100.000 x g a více. Liší se od běžných centrifug zejména tím, že prostor s rotorem musí být při odstřeďování evakuován. Používají se pro dělení biopolymerů a subcelulárních částic v hustotním gradientu sacharosy nebo cesných solí. Tyto techniky lze provádět jak v analytickém, tak i v preparativním měřítku. Analytické centrifugy jsou navíc opatřeny optickým zařízením umožňujícím sledovat rozhraní tvořená jednotlivými sedimentujicími látkami.
Obrázek 8: Nomogram na výpočet relativní odstředívé síly R.
poloměr rotoruRPM xg
RPM počet otáček za minutu (revolutions per minute)xg relativní odstředivé zrychlení (R)poloměr rotoru – vzdálenost osy rotoru od středu dna kyvety (v mm)
Pro většinu prací preparačních i analytických jsou požadovány centrifugy chlazené. Chlazení centrifug je zajišťováno chladicími agregáty, zabudovanými do jediného provozního celku s centrifugou a je ovládáno termostatem. Protilehlé kyvety (u vysokootáčkových centrifug lépe všechny kyvety) musejí být staticky a dynamicky vyváženy. Z bezpečnostních důvodu i z hlediska ochrany centrifugy je nutno klást na vyvažování kyvet zvýšenou pozornost. Každá nepřesnost v dynamickém i statickém vyvážení se mnohonásobně projeví zvětšením odstředivého tlaku na
30

jednu stranu osy. Osa se nerovnovážným zatížením snadno ohne nebo se poškodí ložisko. Při chodu odstředivky se při nepřesném vyvážení projevují silné vibrace. Statického vyvážení se dosáhne tak, že se na technických váhách s přesnosti na jeden gram vyváží protilehlá pouzdra s kyvetami naplněnými suspenzí určenou k centrifugaci. Daleko větší pozornost je třeba klást dynamickému vyvážení protilehlých kyvet. Protilehlé kyvety musí mít stejnou velikost, hmotnost a stejně umístěné těžiště. To znamená, že protilehlé kyvety musí být naplněny stejnou suspenzí, přesněji řečeno suspenzi o stejné hustotě. Nelze tedy například při precipitaci síranem amonným kyvetu s tímto roztokem vyvážit kyvetou s destilovanou vodou.
DIALÝZA
Tato separační technika využívá difúze nízkomolekulárních látek membránou nepropustnou pro velké molekuly a částice z roztoku o vyšší koncentraci do roztoku o koncentraci nižší. V okamžiku, kdy se koncentrace látek procházejících membránou na obou stranách membrány vyrovnají, proces se zastaví. Molekuly proteinů neprocházejí pro svou velikost otvory dialyzačních membrán. Dialýza umožňuje v biochemii např. snadné oddělení solí od roztoků proteinů (při vysolování proteinů síranem amonným), uplatňuje se i při krystalizaci proteinů. Nejčastěji se využívá tam, kde chceme vysokomolekulární látky zbavit nízkomolekulárních nečistot.
Dříve se používaly pro dialýzu membrány typu: kolodium, celofán, pergamen anebo i membrány zvířecího původu (střeva, vaječná blanka, různé měchýře). Nyní jsou komerčně dostupné umělé membrány s vhodnou velikosti pórů ve tvaru dlouhé trubice různého průměru, běžným laboratorním slangem nazývané dialyzační střeva. Potřebný díl dialyzační trubice se odstřihne a na jednom konci pevně zaváže nebo uzavře speciální svorkou. Před naplněním novou trubici vždy pořádně propláchneme a necháme chvíli namočenou v destilované vodě, je třeba vymýt glycerol, kterým jsou trubice impregnovány. Po naplnění preparátem určeným k dialýze se uzavře i druhý konec trubice a vloží se do nádoby s vodou nebo pufrem o nízké iontové síle. Snažíme se, aby dialýza byla co nejrychlejší. Rychlost dialýzy závisí na koncentračním spádu. (zpočátku je rychlejší, postupně se zpomaluje), který je především ovlivněn poměrem objemů vně a uvnitř membrány. Dále rychlost tohoto procesu závisí na teplotě, na počtu a velikostí pórů v membráně, na síle membrány, na elektrických interakcích mezi membránou a difundujícími částicemi a na velikosti plochy membrány. Míchání a obvykle i výměna kapaliny velmi zrychlují postup dialýzy. Protože dialýza trvá zpravidla desítky hodin, provádí se obvykle přes noc při teplotě 0 - 4°C, aby se zabránilo denaturaci proteinů a množení mikroorganismů.
ULTRAFILTRACE
Podobně jako dialýza, používá se ultrafiltrace pro separaci či zakoncentrování rozpuštěných látek s molekulovou hmotností vyšší než 10-4. Ultrafiltrace je proces, při kterém rozpouštědlo a rozpuštěné látky až do určité kritické velikosti procházejí membránou na základě tlakového gradientu (obr. 9), určujícím faktorem je pórovitost membrány. Při ultrafiltraci můžeme využívat buď pozitivní nebo negativní tlak. Pozitivního tlaku dosahujeme inertním plynem, převážně dusíkem a udržujeme ho nad filtrovaným roztokem (retentát). Roztoky se filtrují pod tlakem 0,05 až 0,5 MPa anebo působením odstředivé síly v centrifuze. Naopak negativní tlak udržujeme pomocí vakua, které působí v prostoru již přefiltrovaného roztoku (difuzát). Rozpouštědlo a rozpuštěné látky s menšími relativními molekulovými hmotnostmi než jsou póry v membráně procházejí do ultrafiltrátu. Látky s vyššími molekulovými hmotnostmi se pak nad membránou postupně zahušťují. Zakoncentrovaný retentát můžeme zředit čistým rozpouštědlem a pokračovat v ultrafiltraci. Tím se postupně snižuje podíl nízkomolekulárních látek v retentátu. Při tomto způsobu dialýzy, nazývaném diafiltrace, se požadovaného výsledku dosáhne za 1/10 až 1/100 času potřebného pro konvenční dialýzu.
31

Hlavním požadavkem na membránový filtr je, aby s dostatečnou rychlosti propouštěl rozpouštědlo (vodu) a nízkomolekulární látky a aby jeho póry měly standardní rozměr. Tyto vlastnosti nemají klasické celofánové či celulosové membrány používané pro klasickou filtraci či dialýzu, jejichž stěna má šířku 0,1 – 1,0 mm. Proto se membránové filtry zhotovují z anizotropních polymerů se šířkou stěny 0,1 – 1,5 µm. S takovými filtry by se však těžko manipulovalo, a proto se připevňují na podpůrný materiál, který má obvykle podobné chemické složení a jeho póry jsou podstatně větší než póry ultrafiltru. Průměr pórů membránových filtrů se pohybuje v rozmezí od 0,1 do 50 nm.
Obrázek 9: Zařízení pro ultrafiltraci.
1/ elektromagnetické míchadlo 2/ zásobník na dusík 3/ ocelová láhev s dusíkem 4/ regulátor tlaku 5/ elektromagnetické míchadlo 6/ tlakový zabezpečovací ventil 7/ přívodová hadice dusíku 8/ ultrafiltrační cela 9/ svírací stojan 10/ odtok difuzátu 11/ kontrola rychlosti míchání
V biochemických laboratořích se nejčastěji setkáváme s ultrafiltračními celami a filtry firmy Amicon (obr. 10). Tato firma dodává filtry s póry o průměru 0,2 - 20 nm, které umožňují dělit látky s relativními molekulovými hmotnostmi od 500 do 300.000 Da. Podle typů jsou odolné vůči vodě a různým organickým rozpouštědlům, dá se s nimi pracovat až do teploty 200°C a lze je vystavit působení zředěných kyselin, hydroxidů a oxidačních látek. Při správném zacházení se mohou ultrafiltry používat mnohonásobněkrát za sebou. Pro účinnou ultrafiltraci musíme zajistit, aby se zadržované molekuly nehromadily na povrchu filtru a nezacpávaly jeho póry, toho dosáhneme kontinuálním mícháním nebo probubláváním.
Obrázek 10: Ultrafiltrační cela Amicon 8200.
1/ víko 2/ nádoba na retentát 3/ nosič membrány s odvodnými kanálky 4/ spodní šroub 5/ stojan na celu 6/ magnetické míchadlo 7-8/ těsnění 9-10/ přívod dusíku 11/ odvod difuzátu
32

ÚKOL č. 5: Homogenizace semen ječmene, precipitace jejich proteinů síranem amonným s následnou dialýzou a ultrafiltrací na zařízení Amicon
Nabobtnaná semena obilnin jsou vhodným zdrojem pro studium enzymů aktivních v raných fázích vývoje rostlin, zejména pro vysoké aktivity a snadnou finanční dostupnost. V naší laboratoři se ze semen ječmene a pšenice izoluje cytokinin dehydrogenasa (EC 1.5.99.12), enzym odbourávající rostlinné hormony cytokininy s nenasyceným postranním řetězcem, např. isopentenyladenin či zeatin. Naklíčený ječmen (slad) je také výborným zdrojem amylas, enzymů významných v technologické praxi, především v lihovarnictví a pivovarnictví. Ve sladu jsou zastoupeny dva enzymy, které štěpí α(1-4) glykosidovou vazbu polysacharidů: α-amylasa (1,4-α-D-glukan-glukanonhydrolasa, EC 3.2.1.1) a β-amylasa (1,4-α-D-glukan-maltohydrolasa, EC 3.2.1.2). Jak napovídají systematické názvy, první enzym štěpí molekulu škrobu ve vnitřní části řetězce za vzniku dextrinu (glukanů), kdežto druhý odštěpuje disacharid maltosu z neredukujícího konce řetězce.
MATERIÁL A CHEMIKÁLIE
50 g nabobtnalých ječmenných obilek, 0,1 M Tris/acetátový pufr, pH 7,6, pevný síran amonný, Nesslerovo činidlo na amonné ionty.
Strojek na mletí masa, centrifuga, kyvety, třecí miska s tloučkem, elektromagnetická míchačka, míchadlo, miska s ledem, dialyzační střevo, spona na dialyzační střevo, odměrné válce, ultrafiltrační cela Amicon, ocelová láhev s dusíkem, předvážky, zkumavky, pipety, kádinky, lžička.
POJMY Precipitát - pevná, usazená část po centrifugaci
Supernatant – čirá kapalina nad precipitátem
Difuzát – kapalina procházející membránou při ultrafiltraci
Retentát – roztok vysokomolekulárních látek neprocházející ultrafiltrační membránou a zůstávající v ultrafiltrační cele.
POSTUP
Nabobtnaná semena rozemelte na strojku na maso a navažte přesně 50 gramů rozemleté kaše. Proteiny extrahujte dvojnásobným objemem (100 mL) 0,1 M Tris/acetátového pufru, pH 7,6. Extrakt nechejte za občasného míchání stát 10 minut na ledu. Po deseti minutách extrakt rozdělte do dvou kyvet, které je třeba pečlivě vyvážit na předvážkách. Centrifugujte 20 minut při maximálním možném g (12.000 x g). Supernatant opatrně přelijte do čisté kádinky a odměrným válcem změřte objem. 1/2 mililitru extraktu odeberte do mikrozkumavky a změřte v něm obsah proteinů (navazuje na lab. cvičení č. 5). Podle tabulky 4 zjistěte potřebné množství síranu amonného pro 50% nasycení, toto množství síranu amonného odvážte a důkladně rozetřete ve třecí misce. Do kádinky s extraktem dejte elektromagnetické míchadlo a kádinku umístěte do misky s ledem na elektromagnetickou míchačku. Po dobu 20 minut postupně k extraktu přidávejte odvážené množství síranu amonného, další přídavek vždy až po rozpuštění předešlého. Dokonale rozpuštěný extrakt se síranem amonným rozdělte do dvou kyvet a centrifugujte (12.000 x g, 20 min). Supernatant odstraňte a precipitát rozpusťte v 10 mL 0,02 M Tris/acetátového pufru, pH 7,6, který připravíte naředěním 0,1 M pufru. 5 mL rozpuštěného precipitátu nalijte do předem namočeného dialyzačního střeva a nechejte dialyzovat přes noc proti stejnému pufru v 1 L kádině.
Ze zbylých 5 mL rozpuštěného precipitátu odeberte do mikrozkumavky 0,5 mL (vzorek č. 1) a zbytek umístěte do ultrafiltrační cely Amicon, kterou si předem sestavíte dle návodu. Celu umístěte do misky s ledem na elektromagnetickou míchačku a napojte láhev s dusíkem přes
33

gumovou hadici a redukční ventil. Otevřením láhve s dusíkem se vytvoří v cele tlak, který spustí vlastní ultrafiltraci. Difuzát jímejte do předem připraveného 5 mL odměrného válce. Po odkapání 3 mL difuzátu (vzorek č. 2), uzavřete přívod dusíku, otevřete celu a k retentátu přidejte 20 mL 0,02 M Tris/acetátového pufru, pH 7,6, celu uzavřete a opět spusťte ultrafiltraci. Difuzát jímejte do odměrného válce a po odkapání 18 mL (vzorek č. 3), zopakujte předcházející krok (vzorek č. 4). Po ukončení druhé fáze ultrafiltrace celu otevřete a zbylý retentát opatrně převeďte pomocí pipety do mikrozkumavky (vzorek č. 5), tak aby jste se špičkou pipety nedotkli filtru. Určete objem zahuštěného retentátu a změřte v něm obsah proteinů (navazuje na cvičení č. 5). Celu poté důkladně vypláchněte destilovanou vodou a vraťte vedoucímu cvičení. Nezapomeňte také zkontrolovat redukční ventil láhve s dusíkem, zda je správně uzavřen a zda manometr ventilu ukazuje nulový tlak.
Stanovení amonných iontů reakcí s Nesslerovým činidlem
Princip: Amonné ionty reaguji s Nesslerovým činidlem (tetrajodortuťnatan draselný) za vzniku červenohnědého produktu, který lze stanovit kolorimetricky s maximem při 436 nm.
NH4+ + 2 HgI4
2- + 4 OH- → NH2I.Hg2O + 7 I
- + 3 H2O
Do 7 zkumavek napipetujte 1,5 mL destilované vody, přidejte 0,5 mL vzorku odebraných během ultrafiltrace: 1/ vzorek č. 1, 2/ vzorek č. 2, 3/ vzorek č. 3, 4/ vzorek č. 4, 5/ vzorek č. 5 6/ dialyzační pufr z právě probíhající dialýzy, 7/ čerstvý dialyzační pufr jako blank a nakonec 0,1 mL Nesslerova činidla do každé zkumavky, po 5 minutách okem porovnejte zabarvení v jednotlivých zkumavkách.
VYHODNOCENÍ
1) Popište a zhodnoťte purifikační proces. Srovnejte dva způsoby odsolení proteinů, které jste použili. Využijte výsledků reakce s Nesslerovým činidlem. Navrhněte alternativní kroky, které by jste mohli použít pro homogenizaci, precipitaci a odsolení izolovaných proteinů.
2) Pomocí metody Bradfordové vám vaší kolegové v rámci úlohy Spektrofotometrie stanoví množství proteinů v 10 µL homogenátu (extrakt z obilek po centrifugaci) a zahuštěného vzorku po odsolení v ultrafiltrační cele Amicon. Ze získaných údajů vypočtěte množství proteinů (extrahovatelných) v jednom gramu obilek a procentuálně stanovte obsah proteinů, které vyizolujete z tohoto extraktu po 50% vysolení síranem amonným.
3) Zamyslete se a s pomocí literatury vypište biologicky aktivní látky, které jsou přítomné v obilkách. Zkuste se zamyslet nad jejím kvantitativním obsahem.
Doporučená literatura:
Peč, P. a kol.: Laboratorní cvičení z biochemie, Vydavatelství UP Olomouc, 2000. Káš, J. a kol.: Laboratorní cvičení z biochemie, Nakladatelství Olomouc, 2000. Ferenčík, M. a kol.: Biochemické laboratórne metódy, Alfa Bratislava, 1981. Procházka, S. a kol.: Fyziologie rostlin, Academia Praha, 1998. Dey, P.M. a kol.: Plant Biochemistry, Academia Press London, 1997.
34

Laboratorní cvičení č. 4
EXTRAKCE, FILTRACE, SUBLIMACE A PRÁCE S VAKUOVOU ODPARKOU
EXTRAKCE
Jako extrakce je označováno převedení látky z jedné fáze, v níž je rozpuštěna nebo suspendována, do fáze jiné. Toto převedení je možné, neboť látka se v určitém poměru rozdělí mezi obě fáze. Distribuce rozpuštěné látky mezi dvě kapalné fáze se řídí Nernstovým rozdělovacím zákonem:
ca/cb = K
Podle něj je poměr koncentrací látky ve dvou vzájemně nemísitelných kapalinách při určité teplotě a po dosažení rovnováhy konstantní. K se nazývá rozdělovací koeficient.
Extrakce látky je podle toho snadná tehdy, jestliže je v extrakčním rozpouštědle mnohem lépe rozpustná než v druhé fázi, čili je-li rozdělovací koeficient značně odlišný od jedničky. Pro látky s rozdělovacím koeficientem menším než 100 jediná extrakce nestačí. V takovém případě je nutno extrakci vícekrát opakovat s čerstvým rozpouštědlem.
Pro jednotlivé typy extrakce se historicky vyvinula některá označení, která budou společně se stručnými definicemi uvedena:
Macerace - látka v pevné fázi je za studena opakovaně extrahována dávkami rozpouštědla.
Digesce - látka v pevné fázi je za tepla extrahována opakovanou dávkou rozpouštědla. Nejčastěji tuhou látku zahříváme s rozpouštědlem pod zpětným chladičem a směs poté filtrujeme nebo dekantujeme. Nejčastěji se používá Soxhletův přístroj, v němž je extrakt stále zahušťován. Přístroj pracuje automaticky. Ve varné baňce je rozpouštědlo, později již roztok, které se odpařuje a jeho páry odcházejí širokou trubicí na levé straně středního dílu přístroje (obr. 11) do zpětného chladiče. Zde dochází k jejich kondenzaci. čisté rozpouštědlo stéká do papírové patrony v níž je umístěn extrahovaný materiál. Hladina roztoku ve střední části přístroje neustále stoupá. V okamžiku, kdy její výška dosáhne úrovně horní části tenké přepadové trubičky, dojde k jejímu přetečení zpět do varné baňky. Tak je možno vyextrahovat značné množství látky s malým množstvím rozpouštědla.
Obrázek 11: Soxhletův přístroj
Perkolace - látkou v pevné fázi prosakuje vlastní tíhou rozpouštědlo. Přívodem čistého rozpouštědla je hladina kapaliny v perkolátoru udržována stále ve stejné výši. To zabezpečuje, aby na vyextrahovávaný materiál v horních vrstvách přicházelo stále nové rozpouštědlo (obr. 12).
Vytřepávání – látka je z roztoku extrahována jednou dávkou rozpouštědla nebo opakovaně dalšími dávkami (frakční vytřepávání). Vytřepavání provádíme tak, že vodný roztok nebo méně často suspenzi látky smísíme v dělící nálevce asi z 1/3 až 1/4 objemu extrakčního činidla. Dělící nálevka má být naplněna nejvýše do 2/3 objemu. Uzavřeme ji zábrusovou zátkou a nejdříve
35

protřepeme, přičemž držíme zátku i kohout. Pak otočíme dělící nálevku vypouštěcí stopkou vzhůru a otevřením kohoutu zrušíme vzniklý přetlak. Třepání a odpouštění tlaku opakujeme obvykle 3-4 x. Pracujeme-li s kyselinami, zásadami či jinými žíravinami, používáme ochranné pomůcky! Jednotlivé fáze se většinou oddělí po chvíli stání. Spodní vrstvu vypouštíme kohoutem, horní odléváme hrdlem dělicí nálevky.
Jedním vytřepáním extrakční výtěžky bývají většinou nízké, v optimálním případě odpovídají Nerstovu rozdělovacími zákonu. Provádíme proto extrakci vždy nejméně 3-4 x. Účinnější je vždy opakovaná extrakce menším množstvím rozpouštědla než extrakce celým množstvím najednou.
Nejčastěji používaná extrakční činidla jsou:
Lehčí než voda: diethylether, benzen, hexan
Těžší než voda: methylendichlorid, chloroform, chlorid uhličitý
FILTRACE
Filtrace je oddělování fází pomocí propustného materiálu, který dovoluje průchod pouze jedné z obou fázi. Obyčejně se pod pojmem filtrace rozumí oddělování pevné fáze od kapaliny nebo plynu. Filtrace je v laboratoři velice běžnou operací. Nejčastější je filtrace kapalin, prováděná buď za účelem zbavení kapaliny mechanických nečistot, nebo k izolaci pevné složky, např. při krystalizaci.
Obrázek 12: Perkolátor
Filtrační materiál je určován chemickým charakterem filtrovaného roztoku. Může to být neklížený, tzv. filtrační papír nebo i pórovitá skleněná nebo porcelánová frita, vrstva azbestu nebo skelné vaty apod. Rychlost filtrace závisí na ploše a vlastnostech filtračního prostředí, na počtu a velikosti pórů, na tlaku a teplotě při filtraci, na povaze sraženiny i na viskozitě filtrované kapaliny.
Základní pomůckou při filtraci je filtrační nálevka, do níž se vkládá vhodně složený papírový filtr. Filtrační papír je vyráběn s různou velikostí pórů. Filtr se zhotovuje ze čtverce filtračního papíru složeného pravoúhle na čtvrtiny a sestřiženého do tvaru kruhové výseče. Rozevřením takto upraveného papíru vzniká kuželový filtr, který je z jedné poloviny trojnásobný a z druhé jednoduchý. Tento tzv. hladký filtr filtruje pouze špičkou a poměrně pomalu. Rychleji pracuje filtr skládaný, nazývaný též francouzský. Připraví se z kruhové výseče, která je vějířovitě překládána směrem od středu k obvodu. Při skládání francouzského filtru je nutno dbát, aby při několikerém překládání nedošlo k poškození filtru ve špičce (obr. 13). Je proto nezbytné, aby jednotlivé záhyby neprocházely jedním bodem. Před vložením do nálevky se doporučuje složený filtr rozevřít a obrátit tak, aby původně vnější stěna filtru tvořila vnitřní stěnu. Tím je možné předejít znečištění filtrátu vlákny papíru uvolněnými během skládání a nečistotami z rukou. Skládaný filtr se opírá o stěny nálevky jen hranami, a proto filtruje téměř celou svou plochou na rozdíl od hladkého filtru, který filtruje jen špičkou. Filtrace francouzským filtrem je podstatně rychlejší než filtrace hladkým filtrem téhož průměru.
36
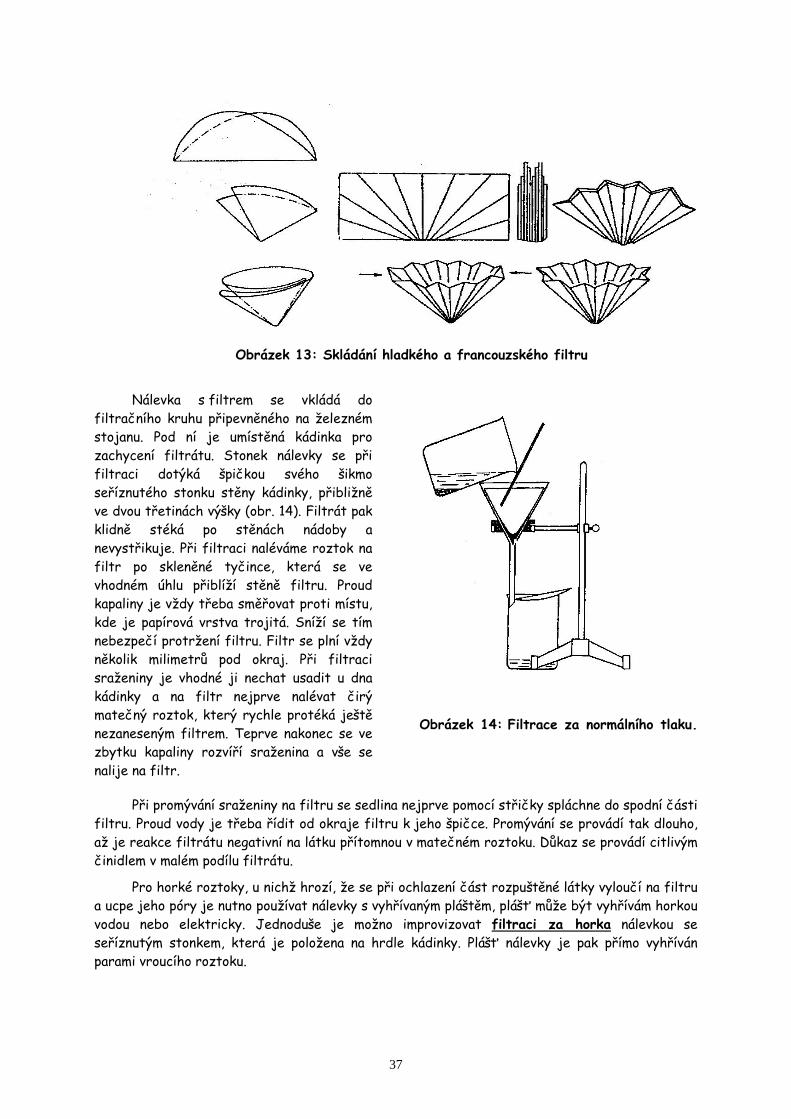
Obrázek 13: Skládání hladkého a francouzského filtru
Nálevka s filtrem se vkládá do
filtračního kruhu připevněného na železném stojanu. Pod ní je umístěná kádinka pro zachycení filtrátu. Stonek nálevky se při filtraci dotýká špičkou svého šikmo seříznutého stonku stěny kádinky, přibližně ve dvou třetinách výšky (obr. 14). Filtrát pak klidně stéká po stěnách nádoby a nevystřikuje. Při filtraci naléváme roztok na filtr po skleněné tyčince, která se ve vhodném úhlu přiblíží stěně filtru. Proud kapaliny je vždy třeba směřovat proti místu, kde je papírová vrstva trojitá. Sníží se tím nebezpečí protržení filtru. Filtr se plní vždy několik milimetrů pod okraj. Při filtraci sraženiny je vhodné ji nechat usadit u dna kádinky a na filtr nejprve nalévat čirý matečný roztok, který rychle protéká ještě nezaneseným filtrem. Teprve nakonec se ve zbytku kapaliny rozvíří sraženina a vše se nalije na filtr.
Obrázek 14: Filtrace za normálního tlaku.
Při promývání sraženiny na filtru se sedlina nejprve pomocí střičky spláchne do spodní části filtru. Proud vody je třeba řídit od okraje filtru k jeho špičce. Promývání se provádí tak dlouho, až je reakce filtrátu negativní na látku přítomnou v matečném roztoku. Důkaz se provádí citlivým činidlem v malém podílu filtrátu.
Pro horké roztoky, u nichž hrozí, že se při ochlazení část rozpuštěné látky vyloučí na filtru a ucpe jeho póry je nutno používat nálevky s vyhřívaným pláštěm, plášť může být vyhřívám horkou vodou nebo elektricky. Jednoduše je možno improvizovat filtraci za horka nálevkou se seříznutým stonkem, která je položena na hrdle kádinky. Plášť nálevky je pak přímo vyhříván parami vroucího roztoku.
37

Podstatně urychlit filtraci je možno použitím sníženého tlaku, odsáváním vzduchu v prostoru pod filtrem. Pro filtraci za sníženého tlaku užíváme Büchnerovy nálevky (obr. 1-17), skleněných nebo porcelánových nučí a filtračních kelímků. Nejednodušší zařízení pro filtraci za sníženého tlaku je Büchnerova nálevka s odsávací baňkou. Dno Büchnerovy nálevky je jemně dírkované, na ně pokládáme filtrační papír tak, aby všechny otvory byly zakryty a papír nepřečníval podél stěn nálevky vzhůru, poněvadž by vzniklými kanálky procházel filtrovaný roztok do odsávací baňky. Nálevka je v odsávací baňce upevněná pryžovou vložkou nebo zátkou. Mezi odsávací baňku a vývěvu je vhodné umístit pojistnou láhev (prázdnou promývačku), která zabraňuje vniknutí vody z vývěvy do odsávací baňky nebo roztoku do vývěvy. Vniknutí vody do odsávací baňky zabráníme, odpojíme-li baňku od vývěvy před zastavením toku vody ve vodní vývěvě.
Při filtraci látek reagujících s filtračním papírem (koncentrované kyseliny, KMnO4 a pod.) používáme skleněné frity, kde funkci filtračního papíru zastává do filtračního kelímku vtavená skleněná pórovitá destička. Velikost pórů je odstupňována v řadě S1-S2-S3-S4-S5, velmi malé póry jsou určeny pro filtraci biologických materiálu. Vzhledem k velké hustotě frit (pomalá filtrace) filtrujeme vždy za použití sníženého tlaku. Nevýhodou skleněných frit je jejich značná cena a hlavně jejich obtížné čištění.
SUBLIMACE
Sublimace je podobně jako krystalizace jednou ze základních metod čištění a oddělování jednotlivých složek směsi. Děj je založen na tom, že tenze par tuhých látek se s rostoucí teplotou zvyšuje, a proto lze mnohé látky převést do plynného skupenství, aniž roztají, a páry opět přímo zkondenzovat v tuhou látku. Bod sublimace je teplota, při níž se tenze par tuhé látky vyrovná s vnějším tlakem. V porovnání s krystalizací má sublimace v mnoha případech značné výhody. Sublimovaný preparát totiž neobsahuje mechanické nečistoty, které i při pečlivé krystalizaci mohou doprovázet výsledný produkt. Při sublimaci jednoduchých směsí se obyčejně pracuje s malými ztrátami a jednoduchá sublimace často nahradí i několikrát opakovanou krystalizaci. Vhodnost použití sublimace jako čistící a separační techniky záleží na vlastnostech dané látky. Rozmezí teplot a tlaků, za nichž lze sublimaci použít je možno zjistit ze stavového diagramu příslušné látky (viz fyzikální chemie).
Zařízení a způsob sublimace se řídí podle toho, jaké vlastnosti má mít sublimát. Čím nižší je teplota chlazeného prostoru, v němž páry kondenzují, tím jemnější krystaly se vytvářejí. Vyšší kondenzační teploty podporují naopak vznik větších a vyvinutějších krystalů.
Nejjednodušším zařízením pro provedení sublimace s nepříliš nízkou sublimační teplotou mohou být dvě zabroušená hodinová skla (viz obr. 15-1). Na spodní sklo umístíme sublimovanou látku, přikryjeme druhým sklem a opatrně zahříváme. Sublimující látka se usazuje na horním hodinovém skle. V případě, že sublimovaná látka padá do surového produktu, můžeme mezi skla položit perforovaný filtrační papír, na němž se sublimovaná látka zachytí. Jiným jednoduchým zařízením je porcelánová miska a na ní postavená nálevka (obr. 15-2). Nálevka má mít o něco menší průměr než miska. Stonek nálevky opatříme volnou vatovou ucpávkou. Mezi misku a nálevku opět vkládáme perforovaný filtrační papír. V případě, že sublimační teplota je nízká, použijeme pro sublimaci aparaturu znázorněnou na obrázku 15-3. Surový produkt nasypeme na dno suché kádinky, kterou uzavřeme baňkou. Do baňky zavádíme až ke dnu stále čerstvou chladící vodu. Sublimující látka se pak usazuje na kulatém dně baňky. Po skončení sublimace baňku opatrně sejmeme a usazené krystaly špachtlí seškrábneme.
38

1 2 3
Obrázek 15: Různé způsoby sublimace
Látky při normálním tlaku nesublimující nebo sublimující jen pomalu, případně látky, které se při sublimaci za vyšší teploty rozkládají, lze často sublimovat při sníženém tlaku. K tomuto účelu používáme aparatury podle obrázku 16.
ODPAŘOVÁNÍ VE VAKUU
Odpařování ve vakuu je v biochemické laboratoři jeden z nejčastějších způsobu odpařování rozpouštědla z roztoků látek. Dá se použít na roztoky jak nízko- tak vysokomolekulárních látek, jejichž objem může být od několika mililitrů až po litry a využívá se při něm efektu snížení bodu varu při sníženém tlaku v systému.
Odpařování malých objemů do 2 mL se dělá v evakuovaném exikátoru nad vhodným sorbentem vody (P2O5, NaOH, CaCl2). Vzorek se umístí do mikrozkumavky, a nebo jiné vhodné nádoby, které jsou v exikátoru uloženy tak, aby vodní páry měly k sušidlu co nejkratší cestu.
Obrázek 16: Aparatura pro sublimaci za sníženého tlaku
Hlavní výhodou této metody je, že je levná a dostupná v každé laboratoři, nevýhodou je dlouhý čas (řádově hodiny) a nemožnost odpařit jiné rozpouštědlo než vodu. Nevýhody se odstraňují přístroji pracujícími na podobném principu, kde se páry rozpouštědla neustále odsávají. Při vakuovém zahuštování roztoků, kde je použito organické rozpouštědlo nastává utajený var, což může často způsobit napěnění vzorku či jeho vystřiknutí. Tyto problémy se dají odstranit odpařováním vzorku za současné centrifugace. Zařízení se obvykle skládá z centrifugy, která může být vyhřívaná, z vymrazovací části a z vakuové pumpy.
Nejběžnější způsob odpařovaní středních objemů je odpařování na vakuové rotační odparce. Rotační vakuová odparka se skládá z odpařovací baňky, pohonné jednotky a z vhodného chladiče s kondenzační baňkou a musí být doplněná zdrojem vakua (vodní vývěva, membránová vývěva). Odpařovací baňka se většinou během odpařování ponoří do vodní lázně, kde je vyhřívána na zvolenou teplotu. Vakuum vytvořené v prostoru s roztokem snižuje její bod varu a rotační pohyb baňky způsobuje intenzivní míchání odpařované směsi spojené se stálým obnovováním filmu na kapaliny na vnitřním povrchu baňky. Tímto se zamezuje přehřívání kapaliny, zvyšuje se odpařovací povrch a zkracuje dráha bublin páry z vnitřního prostoru baňky na povrch roztoku. Důsledkem je intenzivní odpařování kapaliny, jejíž páry kondenzují ve vodním chladiči, umístěném mezi zdrojem vakua a odpařovací baňkou, a stékají do kondenzační baňky. Schéma vakuové odparky je na znázorněno na obrázku 17.
39

Obrázek 17: Schéma vakuové rotační odparky: 1 – odpařovací baňka, 2 – pohonná jednotka, 3 - vodní chladič, 4 – kondenzační baňka, 5 – napouštěcí uzávěr.
ÚKOL č. 6: Izolace kofeinu z čaje a jeho přečištění sublimací
Alkaloidy jsou nejpočetnější skupinou rostlinných látek sekundárního metabolismu. Dosud jich bylo izolováno kolem sedmi tisíc. Z chemického hlediska se dají alkaloidy popsat jako organická báze s jedním nebo několika heterocyklickými dusíkovými atomy v molekule. V přírodě se vyskytují ve formě solí s organickými kyselinami. Alkaloidy obsahuje 10 až 20 % všech vyšších rostlin. Byly nalezeny ve všech jejich částech, ale nejvíce jsou přítomné především v semenech, kořenech a kůře stromů. Kofein patří do skupiny purinových alkaloidů (purin je složen ze dvou heterocyklů – pětičlenného a šestičlenného a obsahuje celkem 4 atomy dusíku na molekulu). Molekula kofeinu má navíc dvě oxo-skupiny (xantin) a je třikrát methylovaná. Kofein je nejhojněji zastoupen v semenech kávovníku a kakaovníku a listech čaje. Má povzbudivý účinek na centrální nervovou soustavu a srdeční činnost, podporuje činnost ledvin a zvyšuje tvorbu moči. Stimulační účinek kofeinu, stejně jako theofylinu z čaje, je založen na inhibici enzymu fosfodiesterasy, která odbourává látky mající podobnou strukturu a přenášející nervový vzruch, jako je např. cyklický adenosinmonofosfát.
MATERIÁL A CHEMIKÁLIE
Kádinky, vařič, filtrační papír, nálevka, dělicí nálevka, vakuová odparka, železný stojan s kruhem, pipety, odpařovací miska, vodní lázeň, baňka, korková zátka, skleněné trubice, hadice, mikrozkumavka, špachtle, parafilm, čajové listy, octan olovnatý, ethanol, chloroform, 5% roztok hydroxidu sodného.
POSTUP
Před začátkem práce si zapněte vodní lázeň u vakuové odparky a vodní lázeň v digestoři na odpařování vzorku, aby jste předešli časovým prodlevám.
Na předvážkách odvažte tři gramy čajového listí a povařte v kádince na vařiči 5 minut v 50 mL destilované vody. Mezitím si připravte 20 mL roztoku octanu olovnatého (w/v = 5%), přidejte ho k čajovému odvaru a povařte dalších 5 minut. Připravte si francouzský skládaný filtr a po vychladnutí roztok přefiltrujte. Filtrát odpařte zhruba na 20 mL objemu kapaliny. S použitím dělící nálevky pak proveďte extrakci odparku s 15 mL chloroformu třikrát po sobě. Spojené extrakty v organickém rozpouštědle promyjte v dělící nálevce destilovanou vodou (15 mL) a potom stejným objemem roztoku hydroxidu sodného (w/v = 5%). Přečištěný chloroformový extrakt zahustěte na vakuové odparce. Postupujte následovně:
40

1) Extrakt přelijte do 250 mL baňky s kulatým dnem a zábrusovým hrdlem. Nezapomeňte vložit varné kuličky. Baňku připevněte svorkou na levé ústí vakuové odparky.
2) Zapněte vodní lázeň na 60°C a pomoci šroubu umístěte baňku do polohy, kdy je její spodní stěna omývána vodou ve vodní lázni.
3) Uzavřete tlačkou hadici na pravé straně vakuové odparky.
4) Do chladiče pusťte mírným proudem vodu a zkontrolujte, zda-li je utěsněna kondenzační baňka.
5) Spusťte vývěvu a evakuujte odparku.
6) Spusťte rotaci baňky.
7) Po skončení odpařování nejprve uvolněte tlačku na pravé trubici odparky a teprve pak vypněte vývěvu.
Odparek rozpusťte v etanolu a baňku kvantitativně etanolem vypláchněte. Ethanolický roztok odpařte na vodní lázni v odpařovací misce.
Připravte si aparaturu k sublimaci podle obrázku 15-3. Odparek kvantitativně převeďte na dno kádinky aparatury. Rozhraní mezi chladicí baňkou a kádinkou utěsněte parafilmem. Aparaturu zahřívejte na vařiči s azbestovou síťkou až do doby, kdy skončí vývoj sublimujících par. Nezapomeňte pustit vodu. Po skončení sublimace zastavte vodu a oddělejte zátku s trubicemi. Krystalky kofeinu vyloučené na dně baňky se pokuste pomocí špachtle kvantitativně převést do předem zvážené mikrozkumavky.
VYHODNOCENÍ
1) Popište a zhodnoťte postup izolace. Navrhněte jiné metody, kterými by jste mohli nahradit jednotlivé kroky.
2) Uveďte výtěžek izolace kofeinu a navrhněte způsob, jak by jste určili jeho čistotu.
3) Za pomocí literatury navrhněte analytickou reakci, kterou by jste dokázali kofein.
Doporučená literatura:
Ferenčík, M. a kol.: Biochemické laboratórne metódy, Alfa Bratislava, 1981. Eysseltová, J. a kol.: Základy laboratorní techniky, Nakladatelství Karolinium Praha, 2000. Kašpárek, F. a kol.: Cvičení z laboratorní techniky, Vydavatelství UP Olomouc, 2000. Čtrnáctová, H. a kol.: Didaktika a technika chemických pokusů, Vydavatelství Karolinium Praha, 1992.
41

Laboratorní cvičení č. 5
SPEKTROFOTOMETRICKÁ MĚŘENÍ
Viditelné záření tvoří malý úsek z oblasti elektromagnetického vlnění v rozmezí délek 400-700 nm. Přilehlá oblast rozmezí vlnových délek 200-400 nm se nazývá blízká ultrafialová oblast, 700-2000 nm se pak nazývá blízká infračervená oblast. Celá oblast vlnových délek se také nazývá oblastí elektronových spekter.
Prakticky v každé biologicky zaměřené laboratoři se dnes setkáváme s přístroji pro měření spektrálních veličin ve viditelné a ultrafialové oblasti. Tzv. spektrofotometry, pro registraci spekter v oblasti od 200 (někdy 185 nm) do 700 nebo 1000 nm. Není divu, vždyť téměř všechny biologicky zajímavé látky (kromě sacharidů) mají v této oblasti, kterou označujeme zkratkou UV-VIS (ultraviolet - visible), charakteristické absorpční pásy a jejich koncentraci lze proto většinou poměrně snadno stanovit na základě jejich spektrálních vlastností.
Určování koncentrace roztoků není jedinou možností využití absorpční spektrofotometrie. Již po několik desetiletí je známo, že absorpční vlastnosti závisejí na interakci příslušného chromoforu s okolím. Z toho vyplývá, že vlnová délka absorpčního maxima i intensita absorpce mohou být ovlivněny řadou procesů, které biochemika enormně zajímají. Uveďme zde pro ilustraci několik příkladů:
- Při denaturaci biopolymeru (bílkoviny nebo nukleové kyseliny) se mění jeho absorpční spektrum, protože chromofory se dostávají do jiného mikroprostředí (u bílkovin zevnitř globule do vodného prostředí, v DNA z jádra helixu, kde těsně interagují se sousedy vodíkovými a patrovými interakcemi).
- Protože fenolátový ion má jiné vlastnosti než nedisociovaný fenol, je možné ze změny spekter bílkoviny při zvyšování pH roztoku určit titrační křivku tyrosinových zbytků a z ní pak usoudit na jejich lokalizaci v molekule proteinu.
- Při nekovalentní vazbě různých ligandů na biopolymer se často mění jejich spektrum; to dovoluje touto metodou určit počet navázaných ligandů a asociační konstantu.
ODVOZENÍ LAMBERT - BEEROVA ZÁKONA
Interakcí záření s hmotou, tj. s atomy a molekulami, dochází k jeho absorpci, tj. k přijetí kvanta energie a zvýšení energie atomu (molekuly) z původního základního stavu do stavu exitovaného. Při absorpci záření v oblasti 200-2000 nm dochází převážně k exitaci elektronového systému, resp. valenčních elektronů zúčastněných atomů a molekul. Vlastní interakce záření s hmotou se sleduje na absorpčním spektru, tj. na záznamu závislostí množství absorbovaného záření na jeho vlnové délce.
Předpokládejme, že na spekrofotometrickou kyvetu s optickou drahou I, která obsahuje absorbující látku, dopadá světlo o intensitě Io. Pokles intensity světla dI v důsledku jeho absorpce vrstvou roztoku o tloušťce dl, pak bude
-dI = k.I.dl
kde k je konstanta úměrnosti. Po separaci proměnných a integraci (interval 0 - 1) získáme vztah
j.c
IIlog o =
42

zvaný Lambertův zákon, kde I je intensita světla vystupujícího z kyvety.
Analogicky můžeme postupovat při studiu závislosti poklesu intensity světla na koncentraci absorbující látky při konstantní délce kyvety. Zde vycházíme z předpokladu, že dI bude, přes jinou konstantu j, úměrné vzrůstu koncentrace dc, tedy:
-dI = j.I.dc
Opět integrujeme, tentokrát v intervalu 0 až c, a získáme Beerův zákon:
j.c
IIlog o =
Zaměřme se nyní na veličiny na levé straně obou rovnic. Veličina Io je intensita světla, které do kyvety vstupuje, a I je intensita světla vystupujícího. Nabízí se vyjádřit poměr těchto hodnot jako frakci světla pohlceného v kyvetě; tato veličina se pak nazývá transmitance T:
T =
I
Io a často se vyjadřuje v procentech. Z fyzikálního pohledu není přirozenějšího popisu absorpčních efektů než pomocí této veličiny. Ta však byla v posledních letech téměř opuštěna, neboť pro praktické laboratorní používání trpí zásadním nedostatkem: není přímo úměrná koncentraci absorbující látky. Přímou úměru zajišťuje veličina na levé straně obou výše uvedených vztahů, která se nazývá absorbance A; tedy
A = log I
I1T
o = log
Dříve se místo ”absorbance” užívalo termínu ”extinkce” (označení E); v anglosaské, zejména americké literatuře, toto označení tvrdoší ně přežívá. Evropský čtenář by si měl být jist, že pojmy absorbance a extinkce (a podobně i absorpční a extinkční koeficient) jsou naprosto ekvivalentní.
j
Poněkud jinak bychom měli hledět na pojem optická hustota, zkracovaný O.D. nebo OD (optical density). Prochází-li paprsek kyvetou, může být jeho intensita snižována i jinými jevy než absorpcí (pohlcením fotonu spojeným s excitací molekuly do vyššího energetického stavu). Pro zeslabení intensity v důsledku absorpce je vhodný termín absorbance, zde také většinou platí, jak uvidíme dále, Beerův zákon. Pokud však je intensita paprsku zeslabována například rozptylem záření na velikých částicích (agregátech molekul při turbidimetrických měřeních, buňkách při proměřování růstových křivek v mikrobiologii apod.), je vhodné toto zeslabení kvantifikovat jako optickou hustotu. Ta je definována stejně jako absorbance, tedy
OD = log I
Io
,
ale nemá přímou souvislost s jevem absorpce. Jinými slovy, optická hustota je pojmem nadřazeným, kdy jenom v některých případech je totožná s absorbancí.
Spojíme-li oba výše uvedené zákony, získáme jeden z nejznámějších fyzikálně-chemických vztahů, zvaný Lambert-Beerův zákon:
43

A = ε.c.l
kde ε je konstanta úměrnosti, odvoditelná z výše definovaných konstant k a j. Jinými slovy, je to absorbance roztoku o jednotkové koncentraci absorbující látky v kyvetě o jednotkové délce. Vzhledem k tomu, že, jak vyplývá z definiční rovnice, je absorbance bezrozměrná veličina, musí mít ε rozměr (délka-1. koncentrace-1). Za jednotkovou délku zde prakticky vždy považujeme 1 cm. Podle toho, v jakých jednotkách dosazujeme koncentrace, nabývá pak ε různých rozměrů.
Pokud je koncentrace vyjádřena v jednotkách mol.l-1, má ε rozměr cm-1.l.mol-1 a hovoříme o molárním absorpčním (postaru extinkčním) koeficientu. Jeho rozměr lze dále upravit vykrácením délkových jednotek, čímž získáme tvar cm2.mmol-1; je dobré si uvědomit, že tento rozměr je zcela totožný s výše uvedeným základním tvarem a mmol nemá nic do činění s nějakou milimolární koncentrací. Jiným, zcela ekvivalentním rozměrem, je M-1.cm-1, kde M označuje molaritu; tento dokonale pochopitelný tvar je užíván zejména v anglosaské literatuře.
Až doposud jsme uvažovali o přítomnosti jediné absorbující látky ve zkoumaném roztoku. Tento stav je však u reálných vzorků spíše výjimečný. Většinou je přítomno několik látek, které absorbují při dané vlnové délce. Měřená absorbance je pak součtem všech dílčích absorbancí.
URČOVÁNÍ KONCENTRACE ROZTOKŮ POMOCÍ ABSORPČNÍ SPEKTROFOTOMETRIE
Jak je patrno z definice absorbance, při jejím měření potřebujeme znát intensity (nebo lépe světelné toky) dvou paprsků: tzv. referenčního (srovnávacího - Io), jenž nebyl oslaben absorpcí, a měrného (I), který prošel kyvetou se vzorkem. Podle způsobu měření dělíme spektrofotometry na několik základních typů:
1) Jednopaprskové přístroje mají jedinou optickou dráhu. Paprsku se nejdříve postaví do cesty referenční vzorek (v laboratorní mluvě zvaný blank). Množství světla, jež jím prošlo, je zaregistrováno jako Io. Poté je do optické dráhy umístěn měřený vzorek, zaregistruje se I a z obou údajů je vypočtena absorbance. Na starších a jednodušších přístrojích tohoto typu se nastavují dva parametry: při zacloněném paprsku se kompensuje tzv. temný tok, který detekční zařízení poskytuje i v situaci, kdy na něj žádné světlo nedopadá (označení "0" souvisí s nulovou transmitancí při zacloněném paprsku), poté se do paprsku umístí referenční vzorek, jehož absorbance je z definice nulová (transmitance 100%). Toto nastavení je nutno provádět pro každou vlnovou délku, a proto se přístroje tohoto typu nepoužívají pro měření spekter. U moderních přístrojů se závislost Io na vlnové délce pro referenční vzorek uloží do paměti a po změření této závislosti pro vzorek (I) se vypočtou hodnoty absorbancí pro všechny vlnové délky. Princip jednopaprskového spektrofotometru je na obrázku 18.
2) Dvoupaprskové přístroje mají oddělené dráhy pro referenční paprsek a pro vzorek. V každém okamžiku proto mohou registrovat I i Io a vypočítat absorbanci. Tyto přístroje, obvykle poněkud dražší, dovolují velmi snadno měřit různé typy diferenčních spekter a mívají i lepší optické parametry (monochromatičnost světla).
3) Pro úplnost se zmiňme o nejnovějším typu spektrofotometrů, tzv. diod-array-detectors (DAD). Tato koncepce se objevila v polovině 80. let. Ostatní spektrofotometry mají monochromátor umístěn před kyvetovým prostorem, takže vzorkem prochází pouze vybrané ("monochromatické") světlo, které je pak přímo zpracováno detektorem. V DAD přístrojích prochází všechno světlo (polychromatické) vzorkem a teprve poté je rozloženo mřížkou; řada světlocitlivých diod pak analyzuje množství světla jednotlivých vlnových délek, které prošlo vzorkem, respektive referenčním roztokem. Toto experimentální uspořádání má sice poněkud nižší kvalitu optických parametrů (rozlišovací schopnost z hlediska spektrální čistoty), jeho
44

předností je však fantastická rychlost měření, neboť intensita světla všech vlnových délek se snímá v jediném okamžiku. Proto je toto uspořádání vhodné zejména pro kinetická měření nebo jako detektory chromatografických zařízení (HPLC).
lampa
čočka
kyvetase vzorkem
monochromátordetektor
zesilovač výstup
Obrázek 18: Princip jednopaprskového spektrofotometru.
Při stanovení koncentrace pomocí měření absorbance se setkáváme s řadou úskalí. Dále probereme nejběžnější příčiny, jež vedou k chybám.
Prvořadým úkolem je proměřit závislost absorbance na koncentraci absorbující látky. Můžeme volit kteroukoli vlnovou délku, až na výjimky však volíme vlnovou délku absorpčního maxima stanovované látky, a to ze dvou důvodů: je zde nejvyšší citlivost stanovení a přesnost stanovení zde nejméně závisí na přesnosti nastavení vlnové délky. Závislost absorbance na koncentraci by měla být přímková a procházet počátkem. Směrnice této přímky je rovna absorpčnímu koeficientu. Setkáváme se však často s odchylkami od této nejjednodušší závislosti.
− Přímka neprochází počátkem. Taková situace je vždy artefaktem: při nulové koncentraci látky musí být absorbance nulová. Dosti obvyklou chybou je to, že při počítačovém vyhodnocení je zvolen nevhodný přímkový model, povolující nenulový úsek na ose y. Pokud přímku "nelze přinutit", aby procházela počátkem, bývá chyba v nevhodné volbě referenčního roztoku, nečisté referenční kyvetě apod.
− Závislost není přímková. K tomuto jevu dochází v případě, kdy se látka vyskytuje v několika spektrálně odlišitelných stavech, přičemž koncentrace jednotlivých složek roztoku závisí na celkové koncentraci. Jako typický příklad se uvádějí oligomerní rovnováhy, kdy oligomer má jiné absorpční spektrum než monomerní jednotky. V těchto případech, kdy neplatí Beerův zákon, nelze snadno určit koncentraci z měření při jediné vlnové délce.
− Pokud látka fluoreskuje, je část absorbovaného světla opět vyzářena. Světlo při vyšších hodnotách absorbance zdánlivě snižuje její hodnotu. Závislost absorbance na koncentraci je pak zakřivená a limituje k nějaké hodnotě.
Při stanovování koncentrace skupiny látek bývá problémem zvolit vhodný "obecný" standard. Typickým příkladem je zde stanovování koncentrace proteinů. Aromatické postranní řetězce bílkovin absorbují v UV oblasti v okolí 280 nm. Často se volí jako standard hovězí sérový albumin, jehož A jednoprocentního roztoku činí 6.8. Zastoupení aromatických aminokyselin v albuminu je však ve srovnání s většinou bílkovin relativně nízké a hodnota jeho specifického absorpčního koeficientu je v "rodině" rozpustných bílkovin nepoužitelná. Na obrázku 19 je srovnání spekter albuminu a imunoglobulinu při stejné koncentraci.
45
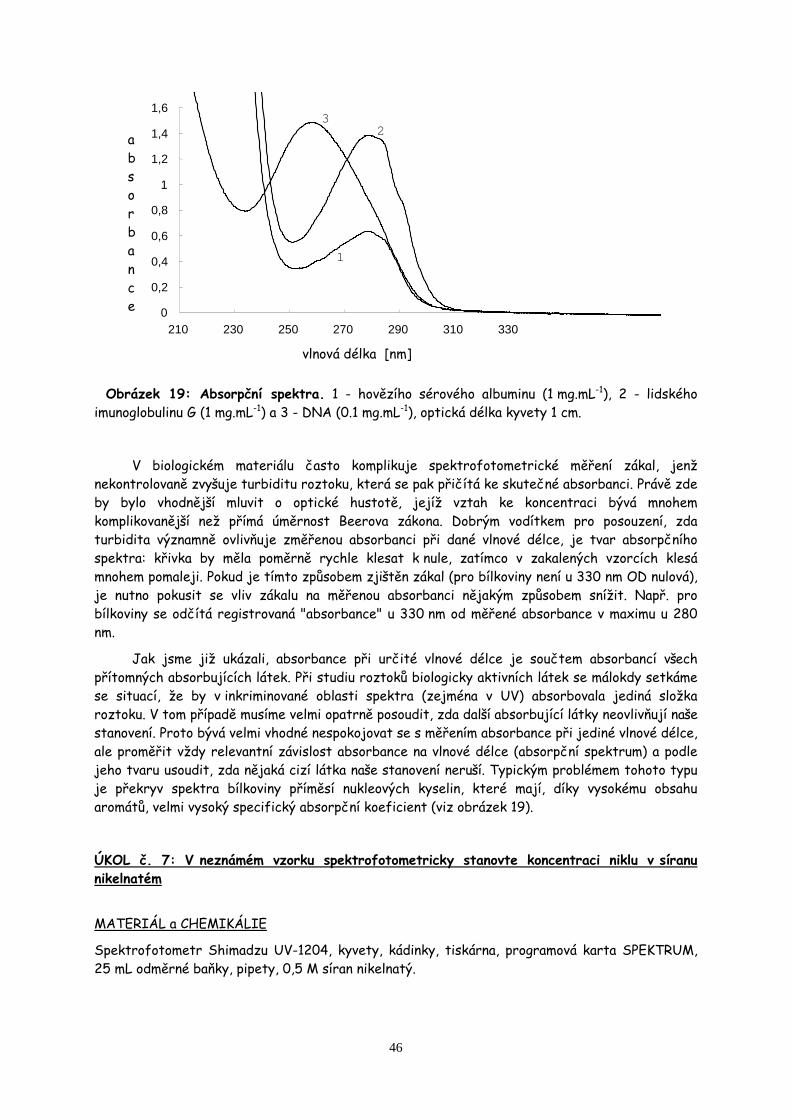
0
0,2
0,4
0,6
0,8
1
1,2
1,4
1,6
210 230 250 270 290 310 330
vlnová délka [nm]
absorbance
1
23
Obrázek 19: Absorpční spektra. 1 - hovězího sérového albuminu (1 mg.mL-1), 2 - lidského
imunoglobulinu G (1 mg.mL-1) a 3 - DNA (0.1 mg.mL-1), optická délka kyvety 1 cm.
V biologickém materiálu často komplikuje spektrofotometrické měření zákal, jenž nekontrolovaně zvyšuje turbiditu roztoku, která se pak přičítá ke skutečné absorbanci. Právě zde by bylo vhodnější mluvit o optické hustotě, jejíž vztah ke koncentraci bývá mnohem komplikovanější než přímá úměrnost Beerova zákona. Dobrým vodítkem pro posouzení, zda turbidita významně ovlivňuje změřenou absorbanci při dané vlnové délce, je tvar absorpčního spektra: křivka by měla poměrně rychle klesat k nule, zatímco v zakalených vzorcích klesá mnohem pomaleji. Pokud je tímto způsobem zjištěn zákal (pro bílkoviny není u 330 nm OD nulová), je nutno pokusit se vliv zákalu na měřenou absorbanci nějakým způsobem snížit. Např. pro bílkoviny se odčítá registrovaná "absorbance" u 330 nm od měřené absorbance v maximu u 280 nm.
Jak jsme již ukázali, absorbance při určité vlnové délce je součtem absorbancí všech přítomných absorbujících látek. Při studiu roztoků biologicky aktivních látek se málokdy setkáme se situací, že by v inkriminované oblasti spektra (zejména v UV) absorbovala jediná složka roztoku. V tom případě musíme velmi opatrně posoudit, zda další absorbující látky neovlivňují naše stanovení. Proto bývá velmi vhodné nespokojovat se s měřením absorbance při jediné vlnové délce, ale proměřit vždy relevantní závislost absorbance na vlnové délce (absorpční spektrum) a podle jeho tvaru usoudit, zda nějaká cizí látka naše stanovení neruší. Typickým problémem tohoto typu je překryv spektra bílkoviny příměsí nukleových kyselin, které mají, díky vysokému obsahu aromátů, velmi vysoký specifický absorpční koeficient (viz obrázek 19).
ÚKOL č. 7: V neznámém vzorku spektrofotometricky stanovte koncentraci niklu v síranu nikelnatém
MATERIÁL a CHEMIKÁLIE
Spektrofotometr Shimadzu UV-1204, kyvety, kádinky, tiskárna, programová karta SPEKTRUM, 25 mL odměrné baňky, pipety, 0,5 M síran nikelnatý.
46

POSTUP
Připravte si sadu 5 roztoků síranu nikelnatého o koncentracích 0,025; 0,05; 0,1; 0,2 a 0,3 M přesným ředěním zásobního roztoku o koncentraci 0,5 M. Postupujte tak, že do odměrných baněk na 25 mL pipetujte 5 mL automatickou pipetou vypočtené množství zásobního roztoku. Roztoky v baňkách doplňte po značku vodou, uzavřete zátkou a dobře promíchejte.
Naučte se obsluhovat spektrofotometr Shimadzu UV-700. Přístroj zapněte a následující měření provádějte vždy přesně podle návodu na obsluhu přiloženého u přístroje. Případné nejasnosti konzultujte s vedoucím cvičení.
Proměření absorpčního spektra 0,3 M roztoku síranu nikelnatého od 600 nm do 800 nm po 10 nm:
1) Na spektrofotometru nastavte nejnižší vlnovou délku tj. 600 nm.
2) Připravte si kyvetu s blankem (destilovaná voda), vložte ji do kyvetového prostoru a přístroj vynulujte funkcí AUTOZERO.
3) Do kyvetového prostoru vložte novou kyvetu s měřeným roztokem a odečtěte příslušnou absorbanci. Kyvetu plňte vždy do 3/4 objemu tak, aby světelný paprsek procházel měřeným roztokem, ne však plnou, aby se při manipulaci roztok nevyléval do přístroje. Před každým měřením optickou stěnu kyvety otřete vatovým tampónem.
4) Postup zopakujte pro všechny vlnové délky, vždy i s předcházející kalibrací na destilovanou vodu.
5) Na závěr podle návodu a pomocí programové karty SPECTRUM proměřte kontinuálně celé spektrum 0,3 M roztoku od 800-300 nm a to tak, že se funkcí RETURN vrátíte do základní nabídky spektrofotometru a postupujete podle návodu. Získané spektrum vytiskněte. Maxima absorpční křivky zjistíte použitím funkce PEAK.
Získanou závislost absorbance na vlnové délce vyneste do grafu na milimetrový papír a porovnejte s vytištěným kontinuálním spektrem. V místě maximální absorbance odečtěte vlnovou délku, při které budete provádět další měření (porovnejte s automatickým vyhledáním maxima funkcí PEAK).
Sestrojení kalibrační křivky
Na spektrofotometru nastavte získanou maximální vlnovou délku. Přístroj vynulujte na destilovanou vodu a bez další kalibrace proměřte sadu připravených roztoků nikelnatých iontů. Mezi každým měřením kyvetu důkladně propláchněte destilovanou vodou a vysušte, aby nedocházelo k ředění a míchání vzorku. Po kalibraci na vodu měření celé sady ještě 2x zopakujte.
Na závěr dejte do kyvety neznámý vzorek a změřte jeho absorbaci. Pokud absorbance překračuje rozmezí hodnot získaných při kalibračním měření, vzorek vhodně nařeďte destilovanou vodou a měření zopakujte.
VYHODNOCENÍ
U každé koncentrace vypočtěte průměrnou hodnotu ze tří naměřených absorbancí. Získanou závislost absorbance na koncentraci vyneste do grafu na milimetrový papír. Z grafu, tzv. kalibrační křivky, pak jednoduše odečtete koncentraci neznámého vzorku. Pozor, berte v úvahu případné ředění neznámého vzorku.
47

ÚKOL č. 8: Praktická aplikace spektrofotometrie v biochemické laboratoři - stanovení obsahu proteinů v biologickém vzorku metodou Bradfordové s vazbou barviva Coomassie Brilliant blue
Barvivo Coomassie Brilliant blue G250 se váže na proteinové molekuly v kyselém pH dvěma způsoby. Trifenylmethanová skupina se váže na nepolární části proteinu a anion sulfoskupiny na bazické skupiny ve vedlejších řetězcích aminokyselin (arginin a lysin). Po vazbě barviva na proteiny dochází k barevné změně, která je úměrná množství proteinu. Reakce je velmi citlivá u albuminu a řady globulárních proteinů. Stanovení je však rušeno řadou interferujících látek, které jsou známy. Metoda je díky své jednoduchosti a citlivosti široce používána. Jako kalibrační protein se používá hovězí sérový albumin (BSA). Barevný produkt barviva s proteinem vykazuje absorpční maximum při 595 nm.
MATERIÁL a CHEMIKÁLIE
Spektrofotometr, kyvety, pipety, zkumavky, vortex, činidlo Bradfordové, kalibrační křivka na BSA.
POSTUP
Do pěti zkumavek napipetujte po 2 mL činidla Bradfordové. Do první a druhé přidejte po 10 µL extraktu z ječmenných obilek, do třetí a čtvrté přidejte po 2 µL zahuštěného retentátu z vysoleného extraktu (vzorky dodají kolegové dělající cvičení č. 3) a pátou ponechte jako blank. Zkumavky promíchejte na vortexu (přístroj pro rychlé a účinné promíchání zkumavek) a nechejte 5 minut stát při laboratorní teplotě. Na spektrofotometru nastavte vlnovou délku 595 nm. Do kyvetového prostoru umístěte v kyvetě vzorek číslo 5 se samotným činidlem Bradfordové a přístroj vynulujte, poté postupně proměřte všechny čtyři připravené vzorky. Po každém vzorku kyvetu vypláchněte ethanolem, jelikož se barvivo Coomassie Brilliant blue váže na stěny skleněných i plastových kyvet.
VYHODNOCENÍ
Hodnoty absorbanci si zaznamenejte a z kalibrační křivky na BSA (dodá vedoucí cvičení) odečtěte obsah proteinů v daném vzorku. Množství proteinu pak přepočtěte na 1 mL vzorku.
ÚKOL č. 9: Měření absorpčních spekter listových barviv
V listech zelených rostlin se vyskytuje větší množství lipofilních barviv, jejichž charakteristickou vlastností je rozpustnost v tucích a tukových rozpouštědlech. Pro fotosyntézu mají hlavní význam chlorofyly a, b a karotenoidy. Jsou značně citlivé vůči vzdušnému kyslíku a světlu. Každé barvivo má své typické maximum, které můžeme pozorovat na absorpčním spektru.
MATERIÁL a CHEMIKÁLIE
Spektrofotometr Shimadzu UV-1204, kyvety, tiskárna, programová karta SPEKTRUM, třecí miska s tloučkem, špachtle, nálevka, filtr, kádinky, pipety, mořský písek, uhličitan vápenatý, hexan, zmražený špenát.
POSTUP
Odvážte 2 g listu špenátu a rozetřete je v třecí misce s malým množstvím mořského písku a uhličitanu vápenatého (na špičku špachtle), na konec přidejte 5 mL hexanu a znovu rozetřete. Směs přefiltrujte přes malý filtr do kádinky.
Do spektrofotometru vložte programovou kartu SPEKTRUM a nastavte rozsah vlnových délek od 800 do 200 nm, pracujte podle návodu přiloženého u spektrofotometru a nezapomeňte
48

zapnout wolframovou lampu pro měření v UV oblasti. Do křemenné kyvety pro UV měření (dodá vedoucí cvičení) nalijte do 3/4 objemu hexan a spektrofotometr vynulujte funkcí BASECORR. Po té do kyvety nalijte hexanový extrakt a proměřte absorpční spektrum. Pokud některá maxima výrazně převyšují absorbanci nad 1,0 vzorek řeďte a to tak dlouho dokud nedostanete čitelné spektrum. Spektrum vytiskněte a pomocí funkce PEAK odečtěte absorpční maxima spektra.
Poznámka: Pro měření v blízké UV oblasti, tzn. 190-350 nm, se používají kyvety ze speciálního křemenného skla, které na rozdíl od skla obyčejného a plastu UV záření propouští. S těmito kyvetami je však nutno pracovat velmi opatrně, jelikož jejich cena mnohonásobně převyšuje cenu běžných kyvet.
VYHODNOCENÍ
1) Odečtěte maxima na absorpčním spektru a pomocí literatury se pokuste zjistit, která barviva se vám podařilo extrahovat ze špenátu. Na základě absorpčního spektra zkuste taky porovnat vzájemně jejich kvantitativní zastoupení v listech špenátu. Z literatury také nastudujte, která další barviva jsou v listech obsažena a vysvětlete, proč jste nepozorovali jejich maxima na absorpčním spektru.
2) Zamyslete se a s pomocí literatury uveďte několik dalších příkladů využití spektrofoto-metrie v biochemii.
ÚKOL č. 10: Spektrální stanovení koncentrace DNA
Nukleové kyseliny absorbují záření v ultrafialové oblasti, maximum absorbance je při 260
nm. Při stanovení je nutné dbát na čistotu preparátu, protože ruší přítomnost proteinů (280 nm) a aromatických látek. Pokud je DNA preparát čistý, poměr A260/A280 dosahuje hodnoty 1.9. Koncentraci DNA lze získat podle vztahu:
cDNA (µg/ml) = 62.9 * A260 – 36.0 * A280 POSTUP
Do 1 cm křemenné kyvety napipetujeme vhodně zředěnou DNA (10-100x). Vzorek DNA dodá vedoucí cvičení. Měříme absorpční spektra vzorku v UV oblasti v rozsahu 200 - 340 nm proti TE pufru a odečteme absorbance při 280 a 260 nm.
VYHODNOCENÍ
Pomocí výše uvedeného vzorce určete koncentraci a čistotu neznámého vzorku DNA.
Doporučená literatura:
Káš, J. a kol.: Laboratorní cvičení z biochemie, Nakladatelství Olomouc, 2000. Eysseltová, J. a kol.: Základy laboratorní techniky, Nakladatelství Karolinium Praha, 2000. Procházka, S. a kol.: Fyziologie rostlin, Academia Praha, 1998. Voet, D. A Voet, J. G.: Biochemie, Victoria Publishing Praha, 1995.
49

Laboratorní cvičení č. 6
DESTILACE
Destilace je metoda užívaná k dělení a čištění směsi kapalin, které se navzájem liší bodem varu. Všeobecně platí, že těkavější složky směsi přecházejí v páry snadněji, takže plynná fáze má jiné složení než kapalná směs a destilát (kondenzát) vzniklý kondenzaci těchto par, který bude bohatší na těkavější složku. Chceme-li dosáhnout dokonalého rozdělení směsi dvou kapalin jedinou destilací v jednoduché aparatuře, musí se jejich body varu lišit alespoň o 50°C a destilované látky nesmějí vytvářet azeotropní směs (směs s konstantním bodem varu). Dle povahy kapalné směsi volíme destilační metodu. Je-li kapalina znečištěna pouze malým množstvím těkavých látek, pak ji čistíme jednoduchou destilací nebo pouhým odpařováním. Máme-li směs látek s velmi blízkými body varu, pak používáme tzv. frakční destilaci (neboli rektifikaci). Je-li destilovaná látka termicky málo stabilní a rozkládá se při nižší teplotě, než je její bod varu, pak jsme nucení použít destilace vakuové. Destilace s vodní párou užíváme k oddělení těch látek, které těkají s vodní párou při nižší teplotě než je jejich bod varu.
ODPAŘOVÁNÍ
Odpařování používáme tam, kde nemáme zájem o odpařené rozpouštědlo. Metodu provádíme na porcelánové misce či v kádince. Odpařovací nádobu zahříváme buď na elektrické vodní lázni (při odpařování látek s bodem varu do 100°C), nebo kahanem přes síťku s azbestem (není-li odpařovaná složka hořlavá). Při odpařování hořlavých látek volného plamene nikdy nepoužíváme a z bezpečnostních důvodu těkavou složku raději oddestilujeme. Rychlost odpařování závisí na teplotě (s rostoucí teplotou se zrychluje) a při bodu varu je nebezpečí, že rychle se vyvíjející páry budou strhávat i zahušťovanou složku. Dále závisí na ploše povrchu kapaliny a na rychlosti odtahu par z prostoru nad kapalinou.
A
B
C
D
Obrázek 20: Součásti destilační aparatury. A/ varná baňka, B/ Claisenův nadstavec, C/ chladič, D/ alonž.
DESTILACE ZA NORMÁLNÍHO TLAKU
Je to nejběžnější druh destilace a užíváme ji všude tam, kde kapalina přechází snadno v páry při teplotě, za které se není třeba obávat rozkladu destilované látky. Destilační aparatura (obr. 20) se skládá z varné baňky (nejčastěji se používá frakční baňka). Varnou baňku nevolíme příliš velkou a plníme ji maximálně do 3/4 objemu. Uzavíráme ji přes Claisenův nádstavec vrtanou zátkou, již prochází teploměr. Jelikož má při destilaci směsi vroucí kapalina obvykle vyšší bod varu než kondenzát, je vhodnější neponořovat teploměr do kapaliny, nýbrž odečítat teplotu odváděných par. Páry vysokovroucích látek (s bodem varu nad 150°C) můžeme kondenzovat ve vzdušném chladiči (obr. 21-1), v ostatních případech používáme vodní chladič Liebigův (obr. 21-2). Ještě účinnější je chladič kuličkový (obr. 21-3) nebo spirálový (obr. 21-4), který musíme při sestavování destilační aparatury postavit do šikmé polohy tak, aby se v něm nezdržoval kondenzát. Na konci chladiče upevníme alonž, pomocí níž stéká kondenzát do jímací nádoby (předloha). Velice výhodné jsou aparatury zábrusové, obzvláště při práci s agresivními látkami.
50

1 2 3 4
Obrázek 21: Chladiče - 1/vzdušný, 2/ Liebigův, 3/ kuličkový, 4/ spirálový.
Poznámka: V dokonale hladkých nádobách může při zahřívání kapalin dojít k tzv. utajenému varu. Je to metastabilní stav, který vznikne přehřátím kapaliny o několik stupňů nad její bod varu. Náhodný impuls pak vyvolává prudký var, vzkypění a přetečení destilované směsi do předlohy. Abychom zabránili vzniku utajeného varu, vkládáme do varné baňky tzv. varná tělíska (skleněné kuličky, úlomky polévaného porcelánu, drcenou cihlu apod.).
VAKUOVÁ DESTILACE
Destilace za sníženého tlaku užíváme tehdy, chceme-li dělit složky z roztoku, v němž je jedna složka při vyšších teplotách labilní. Abychom zabránili rozkladu, musíme při destilaci snížit bod varu roztoku za použití vakua. Aparaturu pro destilaci za sníženého tlaku sestavujeme obvykle z částí s normalizovanými zábrusy (obr. 22). Zvláště výhodné se jeví použití Claisenova nástavce, který spojuje frakční baňku se zábrusovým chladičem a má nahoře dva otvory. Do jednoho otvoru vkládáme zábrusový teploměr, do druhého skleněnou trubičku protaženou do kapiláry, která zasahuje do destilované kapaliny. Význam kapiláry, která spojuje atmosféru s destilovanou kapalinou je v tom, že se při destilaci udržuje konstantní vakuum a zároveň se zajistí promíchávání vroucí kapaliny zaváděním mírného proudu vzduchu. Proud vzduchu regulujeme kohoutem na horní části skleněné trubice, který může být nahrazen nasazením gumové hadice s tlačkou. Kapilárou se také zabraňuje riziku utajeného varu, který je u vakuové destilace daleko pravděpodobnější. Destilační baňku zahříváme vždy jen na vodní lázni nebo topném hnízdě a nikdy nepoužíváme baňky s plochým dnem, které by nevydržely podtlak. Na varnou baňku s Claisenovým nádstavcem je napojen chladič, který je ukončen vakuovou alonží vedoucí destilát do předlohy. Alonž je napojena na zdroj vakua. Mezi předlohou a zdrojem vakua se často zapojuje pojistná nádoba (promývačka) a manometr. Destilaci za sníženého tlaku provádíme při tlaku 100 - 1000 Pa. Snížení tlaku vede ke snížení bodu varu kapalin. Var kapaliny nastává, jak známo tehdy, když se tenze jejich par rovná atmosférickému tlaku, v tomto případě tlaku v aparatuře. Použitím dobré vodní vývěvy je možno snížit bod varu kapaliny o 80-90°C.
DESTILACE S VODNÍ PAROU
Podle Daltonova zákona je tlak nad směsí dvou nebo několika kapalin roven součtu parciálních tlaků jednotlivých složek. Jelikož var kapaliny nastane v okamžiku, kdy tenze její páry se rovná okolnímu – atmosférickému tlaku, je zřejmé, že bod varu takové směsi bude nižší než body varu jednotlivých oddělených složek. Jako příklad uvedeme pro jednoduchost směs anilinu a vody, která vře při 98°C, když tenze vodní páry činí 9,42.104 Pa a tlak par anilinu je 7,07.103 Pa. Bod varu samotného anilinu pak 184.4°C. Destilace s vodní parou je tedy založena na využití Daltonova zákona. Používá se hlavně v organické chemii. Tímto způsobem je totiž možno předestilovat látky, které mají bod varu i vyšší než 200°C při teplotách nižších než 100°C a uchránit je tak před nežádoucím rozkladem.

Obrázek 22: Vakuová destilace.
Při destilaci s vodní parou používáme aparatury sestavené dle obrázku 23. Aparatura se skládá z vyvíječe vodní páry, jimž je varná baňka uzavřená 2x vrtanou zátkou. Jedním otvorem prochází dlouhá skleněná trubice sahající až ke dnu nádoby. Je to tzv. pojistná trubice a jejím úkolem je zabránit při náhlém poklesu tlaku ve vyvíječi nasátí kapaliny z destilační baňky. Podtlak se vyrovná nasátím vzduchu pojistnou trubicí. Vodní pára pak probublává destilační baňkou, kterou zahříváme souběžně s vyvíječem vodní páry. Destilační baňku většinou upevňujeme vzhledem k ostatní aparatuře do šikmé polohy a to proto, aby kapalina, rozstřikována proudem vodní páry, nevnikla do chladiče a neznečišťovala tak kondenzát.
A
B
C
F
E
D
G
H
Obrázek 23: Aparatura pro destilaci s vodní parou. A/ vyvíječ vodní páry, B/ pojistná trubice, C/ destilační baňka, D/ Claisenův nádstavec E/ chladič, F/ alonž, G/ předloha, H/ místo pro teploměr.
52

ÚKOL č. 11: Izolace hřebíčkové silice z hřebíčku destilací s vodní parou Hřebíčková silice obsahuje vedle malých množství jiných látek 85 – 90% eugenolu a 9 –10% acetyleugenolu. Tyto látky mají teploty varu 249 a 253°C a jsou ve vodě jen velmi málo rozpustné. Při teplotě varu vody však mají značnou tenzi páry a lze je proto z hřebíčku snadno vydestilovat s vodní parou při teplotě nižší než 100°C. Ze získané vodní emulze se silice vyextrahuje do vhodného rozpouštědla. Oddestilováním rozpouštědla z extraktu lze získát čistou hřebíčkovou silici.
Eugenol je fenolická látka obsažená hlavně v nezralých plodech rostlin čeledi Myrtaceaejako je Pimento officinalis či Caryophyllum aromaticum u nás známých jako nové koření (jamajský pepř) a hřebíček. Eugenol se používá v zubním lékařství jako lokální anestetikum. Působí také jako aktivátor žaludečních trávicích enzymů a s tím také souvisí obliba těchto druhů koření téměř ve všech světových kuchyních.
MATERIÁL
Třecí miska s tloučkem, topné hnízdo na 250 mL a 500 mL, vodní lázeň, stojany, svorky, baňky, korkové zátky, skleněné trubice, chladiče, alonže, Erlenmaeyerovy baňky, kadiny, teploměr, dělicí nálevka, odměrný válec, skleněné kuličky, hřebíček, hexan.
POSTUP
Sestavte destilační aparaturu pro destilaci s vodní párou dle obrázku 23. Obě baňky umístěte do topných hnízd. Zábrusy dobře promažte silikonem. Vyvíječ vodní páry naplňte do 2/3 vodou a na jeho dno nezapomeňte dát skleněné kuličky (varné kamínky). Do trojhrdlé baňky nasypte 10 gramů jemně rozdrceného a ve třecí misce rozetřeného hřebíčku a přilijte 100 mL destilované vody. Zaváděcí trubičku z vyvíječe vodní páry zaveďte do trojhrdlé baňky. Mírným proudem pusťte do chladiče vodu. Zapněte nejdříve vyhřívání vyvíječe vodních pár a až začnou do destilační baňky unikat vodní páry spusťte i vyhřívání druhé baňky zhruba na polovinu stupnice. Během destilace neustále kontrolujte aparaturu, případně regulujte teplotu vyhřívání. Suspenzi silice s vodou jímejte do připravené předlohy. Z časových důvodu ukončete destilaci asi po 1/2 hodině i v případě, že veškerá silice nebude z hřebíčku vydestilovaná.
Ze získané směsi vody a silice vyextrahujte silici hexanem. Předestilovanou směs nalijte do dělící nálevky, přidejte k ní 15 mL hexanu a po uzavření nálevky zátkou obsah důkladně protřepte (viz úloha č. 4 – Extrakce). Pozor na vnitřní přetlak, odstraníte ho otočením nálevky a otevřením ventilu. Po důkladném protřepání vyčkejte až se v nálevce vytvoří rozhraní dvou vrstev. Spodní vrstvu tj. vodu se zbytkem silice odlijte do kádinky. Roztok silice v hexanu (horní vrstva) pak přelijte do SUCHÉ Erlenmayerovy baňky. I minimální množství vody způsobí zakalení silice po oddestilování hexanu. Extrakci zopakujte ještě dvakrát vždy s novým podílem hexanu, který přidáváte k vodné fázi. Jednotlivé podíly silice v hexanu spojte a oddestilujte rozpouštědlo. Sestavte aparaturu pro destilaci za normálního tlaku. Roztok silice přelijte do malé zábrusové baňky (100 mL), na kterou nasadíte Claisenův nadstavec s vodním chladičem. Baňku zahřívejte opatrně na vodní lázni (vařič s hrncem). Během destilace je nutno regulovat teplotu vodní lázně. Udržujte ji mezi 80 - 90°C. Zároveň sledujte teplotu v destilační aparatuře. Jakmile přestane hexan destilovat, destilaci zastavte a nechte aparaturu zchladnout. Mikropipetou převeďte silici do předem odvážené mikrozkumavky a určete její objem a hmotnost. Oddestilovaný hexan přelijte do láhve k tomu určené.
Pozor: Hexan je hořlavina I. třídy, nepoužívejte v blízkosti práce s ním otevřeného ohně.
Zabraňte, aby se dostala voda dovnitř topného hnízda. Pokud se tak stane okamžitě odpojte hnízdo z elektrické sítě.
53

VYHODNOCENÍ
1) Určete výtěžek hřebíčkové silice a vypočtěte její hustotu.
2) Stanovte teplotu varu hexanu a srovnejte ji s tabulkovou hodnotou.
3) Zamyslete se nad tím, proč je výhodnější destilaci s vodní parou provádět výše popsaným a provedeným způsobem, než destilovat danou látku společně s vodou v jedné baňce.
4) Do protokolu zakreslete vzorce eugenolu a acetyleugenolu a pokuste se odvodit jejich systematické názvy.
Doporučená literatura:
Eysseltová, J. a kol.: Základy laboratorní techniky, Nakladatelství Karolinium Praha, 2000. Kašpárek, F. a kol.: Cvičení z laboratorní techniky, Vydavatelství UP Olomouc, 2000. Mareček, A. a Růžička, A.: Laboratorní technika pro nechemické obory, Vydavatelství MU Brno, 1997 Chemické analytické tabulky
54

Laboratorní cvičení č. 7
PRÁCE S PLYNY A SKLEM
V současné době jsou téměř všechny plyny dostupné komerčně v tlakových lahvích. Občas si však potřebujeme v laboratoři připravit malé množství plynu sami. Nejjednodušším zařízením k tomuto účelu je Oswaldova baňka (obr. 24). Její princip je patrný z obrázku: k tenkostěnné skleněné baňce je přes korkovou zátku připojena dělící, popř. klasická nálevka. Na dno baňky dáváme tuhou látku, případně suspenzi s vodou, ke které z dělící nálevky přikapáváme vhodnou kapalinu, se kterou tuhá látka reaguje za vzniku žádaného plynného produktu. Uvolněný plyn se odvádí trubičkou. Dokonalejším zařízením je Kippův přístroj (obr. 25), v němž se při přerušení odběru plynu vývoj plynu samovolně zastaví. Do střední části C se na dírkovanou kaučukovou destičku vloží tuhá kusovitá látka. Do horní části D nalijeme tolik kyseliny, aby zaplnila celou spodní část a dostala se do styku s tuhou látkou. V tomto okamžiku se začne uvolňovat plyn. Je-li kohoutek B uzavřený, vzniklý plyn vytlačí kapalinu z části A, čímž přeruší další vývoj plynu. Kohoutkem je tedy možno vývoj plynu regulovat. Horní rezervoár D uzavíráme zátkou, zamezujeme tak unikání nežádoucích plynů do ovzduší.
C
D
Obrázek 24: Oswaldova baňka. Obrázek 25: Kippův přístroj A/ spodní prostor pro
kapalinu, B/ odvaděč plynu, C/ střední prostor pro pevnou látku, D/ zásobník na kapalnou složku reakce.
Laboratorně lze běžné plyny připravit následovně:
1) Reakcí tuhé látky s kapalinou
CO2: kusový mramor + 4 M kyselina chlorovodíková
CaCO3 + 2 HCl → CaCl2 + H2O + CO2
H2S: kusový sulfid železnatý + 3 M kyselina chlorovodíková
FeS + 2 HCl → FeCl2 + H2S
H2: granulový zinek + 4 M kyselina chlorovodíková
Zn + 2 HCl → ZnCl2 + H2
55

Cl2: manganistan draselný + konc. kyselina chlorovodíková
2 KMnO4 + 16 HCl → 2 KCl + 2 MnCl2 + 8 H2O + 5 Cl2
oxid manganičitý + konc. kyselina chlorovodíková
MnO2 + 4 HCl → MnCl2 + 2 H2O + Cl2
chlorové vápno + konc. kyselina chlorovodíková
ClOCl2 + 2 HCl → CaCl2 + H2O + Cl2
SO2: měď + konc. kyselina sírová
Cu + 2 H2SO4 → CuSO4 + H2O + SO2
disiřičitan draselný + 2 M kyselina sírová
K2S2O5 + H2SO4 → K2SO4 + H2O + 2 SO2
C2H2: karbid vapenatý + voda
CaC2 + 2 H2O → Ca(OH)2 + C2H2
O2: katalytický rozklad 30% peroxidu vodíku účinkem burelu
2 H2O2 2 H⎯⎯ →⎯ 2MnO2O + O2
HCl: chlorid sodný + konc. kyselina sírová
2 NaCl + H2SO4 → Na2SO4 + 2 HCl
HN3: chlorid amonný + 50% hydroxid amonný
NH4Cl + KOH → KCl + H2O + NH3
HBr: bromid sodný + 85% kyselina fosforečná
3 NaBr + H3PO4 → Na3PO4 + 3 HBr
2) Reakcí dvou kapalin
O2: 15% peroxid vodíku + nasycený roztok manganistanu draselného v 1 M kys. sírové
2 KMnO4 + 5 H2O2 + 4 H2SO4 → 2 MnSO4 + K2SO4 + 8 H2O + 5 O2
SO2: nasycený roztok hydrogensiřičitanu sodného + 3 M kyselina sírová
NaHSO3 + H2SO4 → NaHSO4 + H2O + SO2
CO: dehydratací kyseliny mravenčí účinkem konc. kyseliny sírové
HCOOH H⎯⎯ →⎯ 42SOH2O + CO
CO2: nasycený roztok hydrogenuhličitanu draselného + 3 M kyselina sírová
KHCO3 + H2SO4 → KHSO4 + H2O + CO2
HCl: dehydratací kyseliny chlorovodíkové účinkem konc. kyseliny sírové
N2: 10% roztok chloridu amonného + 10% roztok dusitanu sodného
NH4Cl + NaNO2 → NaCl + 2 H2O + N2
56
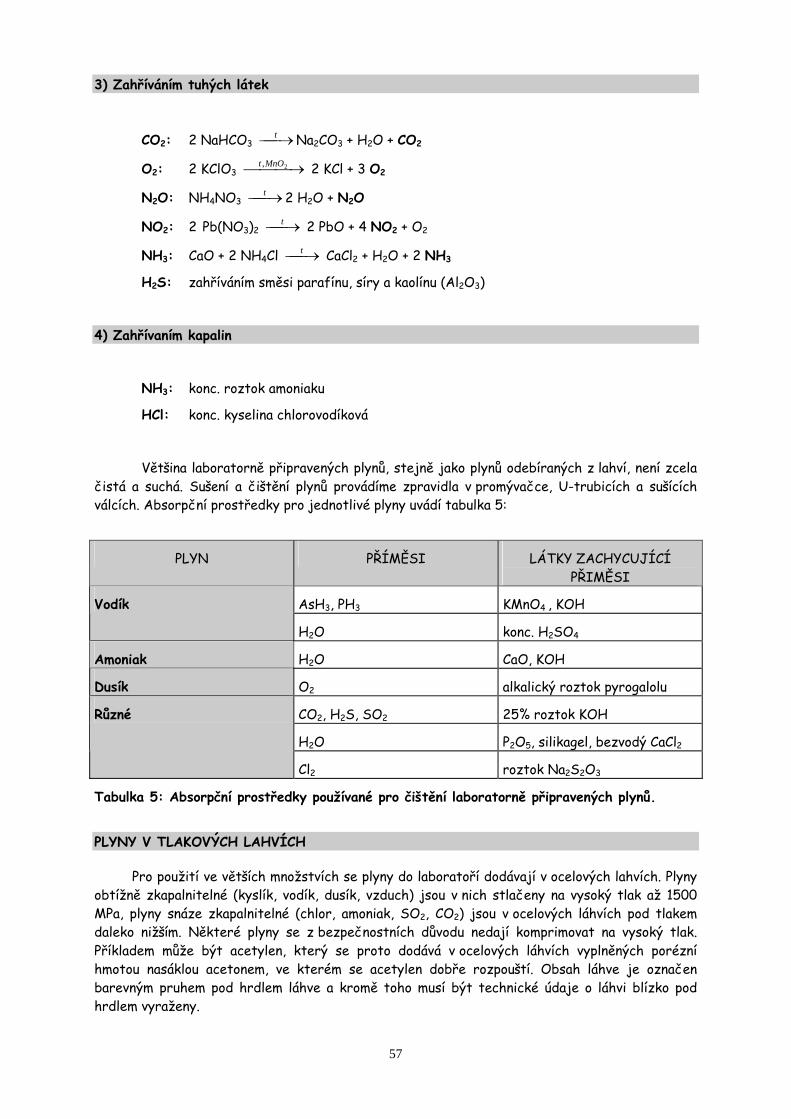
3) Zahříváním tuhých látek
CO2: 2 NaHCO3 Na⎯→⎯t2CO3 + H2O + CO2
O2: 2 KClO3 2 KCl + 3 O⎯⎯ →⎯ 2,MnOt2
N2O: NH4NO3 2 H⎯→⎯t2O + N2O
NO2: 2 Pb(NO3)2 2 PbO + 4 NO⎯→⎯t2 + O2
NH3: CaO + 2 NH4Cl CaCl⎯→⎯t2 + H2O + 2 NH3
H2S: zahříváním směsi parafínu, síry a kaolínu (Al2O3)
4) Zahřívaním kapalin
NH3: konc. roztok amoniaku
HCl: konc. kyselina chlorovodíková
Většina laboratorně připravených plynů, stejně jako plynů odebíraných z lahví, není zcela čistá a suchá. Sušení a čištění plynů provádíme zpravidla v promývačce, U-trubicích a sušících válcích. Absorpční prostředky pro jednotlivé plyny uvádí tabulka 5:
PLYN PŘÍMĚSI LÁTKY ZACHYCUJÍCÍ PŘIMĚSI
AsH3, PH3 KMnO4 , KOH Vodík
H2O konc. H2SO4
Amoniak H2O CaO, KOH
Dusík O2 alkalický roztok pyrogalolu
CO2, H2S, SO2 25% roztok KOH
H2O P2O5, silikagel, bezvodý CaCl2
Různé
Cl2 roztok Na2S2O3
Tabulka 5: Absorpční prostředky používané pro čištění laboratorně připravených plynů.
PLYNY V TLAKOVÝCH LAHVÍCH
Pro použití ve větších množstvích se plyny do laboratoří dodávají v ocelových lahvích. Plyny obtížně zkapalnitelné (kyslík, vodík, dusík, vzduch) jsou v nich stlačeny na vysoký tlak až 1500 MPa, plyny snáze zkapalnitelné (chlor, amoniak, SO2, CO2) jsou v ocelových láhvích pod tlakem daleko nižším. Některé plyny se z bezpečnostních důvodu nedají komprimovat na vysoký tlak. Příkladem může být acetylen, který se proto dodává v ocelových láhvích vyplněných porézní hmotou nasáklou acetonem, ve kterém se acetylen dobře rozpouští. Obsah láhve je označen barevným pruhem pod hrdlem láhve a kromě toho musí být technické údaje o láhvi blízko pod hrdlem vyraženy.
57

Přehled barevných označení tlakových láhví: modrá……………..…kyslík žlutá………………………..chlor zelená……………….dusík fialová..……………….….amoniak červená………….…vodík černá…..………………....oxid uhličitý stříbrošedá……..vzduch perlově šedá…………..oxid siřičitý bílá………………….…acetylen pastelově zelená…..oxid dusný
Kromě barevného označení jsou ventily k láhvím s hořlavými plyny opatřeny levotočivým závitem, ventily k láhvím s nehořlavými plyny mají závit pravotočivý. Ocelové láhve je nutno chránit před zbytečným zahříváním a před nárazem, kdy by skryté vady materiálu a koroze oceli mohly způsobit explozi. V laboratoři je proto nutné uložit láhve do speciálního stojanu nebo připoutat řetězem k pevné opoře.
Odběr plynů z láhví můžeme provádět pouze přes redukční ventily různých typů. Pro plyny dodávané pod vysokým tlakem, používáme nejčastěji redukční ventil zobrazený na obrázku 26. Redukční ventil našroubujeme na vývod láhve pomocí matice. Těsnící kroužek nesmí být poškozený, aby mohl dokonale těsnit. Dříve než otevřeme ventil na láhvi, uzavřeme na doraz uzavírací ventil a odšroubujeme otáčením doleva regulační šroub tak, abychom necítili tlak pružiny. Po otevření ventilu na láhvi ukáže manometr vyžadovaný tlak plynu v láhvi. Zašroubováním šroubu naregulujeme vyžadovaný tlak na manometru. Láhev je takto připravená k odběru plynu. Pomalým otáčením uzavíracího ventilu nastavíme vyžadovaný tlak proudu plynu, přičemž tlak odebíraného plynu se samovolně reguluje prohýbáním membrány a dosednutím čepu do ložiska. Do redukční komory zasahuje pojistný ventil, kterým unikne plyn, když se redukční zařízení pokazí. Redukční ventily, zejména na kyslík, se nesmí nikdy z bezpečnostních důvodů mazat organickými mazadly a ani těsnící složky nesmějí být z organických látek. Užívají se proto kroužky azbestové a olověné.
Obrázek 26: Redukční ventil.
K zachycování a krátkodobému skladování plynů používáme tzv. plynojemy s uzavíracími kapalinami. Nejjednodušším zařízením je zkumavka nebo válec naplněný vodou a ponořený dnem nahoru do větší nádoby s vodou. Plyn zavádíme do zkumavky ohnutou trubičkou. Je-li zkumavka kalibrována, pak mluvíme o tzv. eudiometru (obr. 27), jehož se užívá ke stanovení objemu vzniklého plynu. Větší množství plynu uchováváme ve skleněných nebo kovových plynojemech. Jednoduchý plynojem představují dvě láhve, opatřené dole tubusem a navzájem spojené pryžovou
58

hadicí. V jedné láhvi je v hrdle pryžová zátka s trojcestným kohoutem. Toto zařízení umožňuje nasávání plynu při plnění a zároveň nastavení libovolného přetlaku při odběru plynu. Plyny nasáváme tak, že láhev s trojcestným kohoutem naplníme zcela uzavírací kapalinou. Pak ji pomocí trojcestného kohoutu spojíme s prostorem, v němž se vyvíjí plyn. Jakmile prázdnou láhev snížíme, začne se plnit uzavírací kapalinou, která do ní protéká z láhve opatřené trojcestným kohoutem. Tím nad kapalinou jímáme plyn, který se nasává do prostoru, v němž původně byla kapalina. Po uzavření kohoutu můžeme plyn nad kapalinou uchovávat. Chceme-li plyn odebírat, spojíme nastavením trojcestného kohoutu láhev s prostorem, kam má být plyn transportován a druhou láhev zvedneme nad hladinu láhve s plynem.
ÚKOL č. 12: Práce se sklem a korkem
Naučte se řezat a otavovat skleněné trubičky, ohýbat je, vytahovat z nich kapiláry a zatavovat jejich konce.
Skleněné trubice nebo tyčinky se při řezání položí na stůl a nožem na sklo nebo jemným pilníkem se udělá asi na polovinu obvodu vryp. Trubice se uchopí oběma rukama co nejblíže vrypu tak, aby byly palce opřeny do trubice na opačné straně vrypu. Mírným tahem a současným tlakem se trubice rozlomí. Nedá-li se trubice snadno rozlomit, je nutné vryp prohloubit, neboť by trubice mohla prasknout nepravidelně. Tlustostěnné trubice a trubice o velkém průměru se řežou tak, že se udělá vryp po celém obvodu trubice a na rysku se přiloží za stalého pomalého otáčení silný rozžhavený drát ohnutý do půlkruhu. Ostré hrany trubic je třeba hladce otavit. Zamezí se tak zbytečným úrazům pořezáním, zároveň je tím usnadněno vsazování trubic do pryžových hadic nebo zátek. Konec trubice se otáčí a zahřívá v plameni tak dlouho, až sklo začne měknout, což se projeví jasně žlutou barvou plamene. Pozor, při delším zahřívání by se konec trubice mohl zúžit a nakonec by se úplně uzavřel.
Při ohýbání trubice je důležité rovnoměrné prohřátí trubice po celé délce ohybu. Trubici je třeba držet v plameni podélně a stále ji otáčet a posouvat, aby se dosáhlo rovnoměrného ohřevu. Zahřívá se tak dlouho, až se sklo začne vlastní váhou ohýbat. Po vytažení trubice z plamene je nutno ji ihned ohnout do požadovaného tvaru a sečkat, až sklo dostatečně ztuhne. Plného tvaru ohnutí lze dosáhnout, když se během ohýbání do trubice mírně fouká při utěsnění druhého konce.
Při vytahování kapilár je opět nutné důkladné prohřátí trubice v délce zhruba 6 cm. Když sklo dostatečně změkne, stlačí se oba konce trubice mírně k sobě, aby se na nažhaveném úseku nahromadilo více skloviny. Po vyjmutí trubice z plamene se ihned rychlým tahem vytáhne kapilára do požadované délky a šířky. Po ochlazení skla se kapilára vylomí ze zbytku trubice, popř. odřízne nožem na sklo. Konec kapiláry lze zatavit krátkým podržením v plameni.
Skleněné trubice se mohou spojovat stavováním jen v případě, jsou-li ze stejného skla nebo ze skla podobných vlastností. Při spojování trubic stejného průměru je postup takový, že se jedna z trubic na jednom konci zazátkuje a volné přilehlé konce trubic se protaví v plameni. Mimo plamen se trubice přiloží rozžhavenými konci k sobě a za stálého otáčení se lehce stlačí. Trubice se musí dokonale stavit, a spojení musí být bez nejmenších otvorů. Nahromaděné sklo ve sváru se vyrovná foukáním po předchozím protavení, do otevřeného konce trubice. Při stavování trubic nestejného průměru je potřeba širší trubici vytažením zúžit na průměr menší trubice.
Na vrtání otvorů do korkových a gumových zátek se užívá korkovrtů, což jsou tenkostěnné mosazné trubice na jednom konci naostřené. Korková zátka se nejprve změkčí ponořením na několik minut do vařící vody. Pro vrtání se vybírá vždy korkovrt s o něco menším průměrem než má trubička, která se bude do otvoru vsazovat. Zátku je třeba položit na tvrdou podložku a vrtat kolmo k ploše zátky šroubovitým pohybem. Po provrtání zátky se vyvrtaná hmota odstraní
59

z korkovrtu tyčinkou a korkovrt se vytáhne. Trubičky se do otvorů vsouvají šroubovitým pohybem. Trubičku je lépe držet co nejblíže zátce a z bezpečnostních důvodů nejlépe v hadříku.
MATERIÁL
Skleněná trubice o průměru 6 mm s tenkou stěnou, skleněná trubice o průměru 6 mm s tlustou stěnou, nůž na sklo, podložka, hořák, korková zátka, korkovrt, gumová hadice.
POSTUP
Podle přiložených materiálů se seznamte se základní charakteristikou práce s chemickým sklem a práci s korkovrtem. Naučte se řezat a otavovat skleněné trubičky, ohýbat je, vytahovat z nich kapiláry a zatavovat jejich konce. Pokuste se o vyfouknutí baničky.
Na závěr využijte dovedností, které jste se naučili. Sestavte aparaturu dle obrázku 27, kterou budete potřebovat pro následující úkol. Využijte skleněné trubice se širší stěnou, korkovou zátku, do které pomocí korkovrtu vyvrtáte dva otvory (jeden pro dělící nálevku a druhý pro připravenou odvodovou trubici). Pro lehčí manipulaci s aparaturou tuto trubici připravte dvoudílnou a díly propojte gumovou hadicí.
Obrázek 27: Aparatura pro měření objemů plynu uvolněného chemickou reakcí (eudiometr).
ÚKOL č. 13: Určete procentuální obsah uhličitanu vápenatého ve vaječné skořápce jeho rozložením na oxid uhličitý.
Někteří živočichové a rostliny jsou schopní akumulovat ve svých tělech potravou a živinami přijímaný vápník ve formě uhličitanu. Tato ryze anorganická látka pak díky své pevnosti a tvrdosti plní především roli ochrannou a je vylučována na povrch, např. někteří zástupci měkkýšů nebo u vejcorodých obratlovců chrání vajíčka, kde je hlavní složkou jejich obalů. O přítomnosti uhličitanu vápenatého ve vaječné skořápce se dá přesvědčit jejím rozkladem koncentrovanou kyselinou chlorovodíkovou a jímáním uvolněného oxidu uhličitého dle následující rovnice:
CaCO3 + 2 HCl → CO2 + H2O + CaCl2
Plyn uvolněný během reakce se může jímat pomocí jednoduše zhotoveného eudiometru a z objemu lze vypočítat na základě Avogadrova zákona množství původního CaCO3.
60
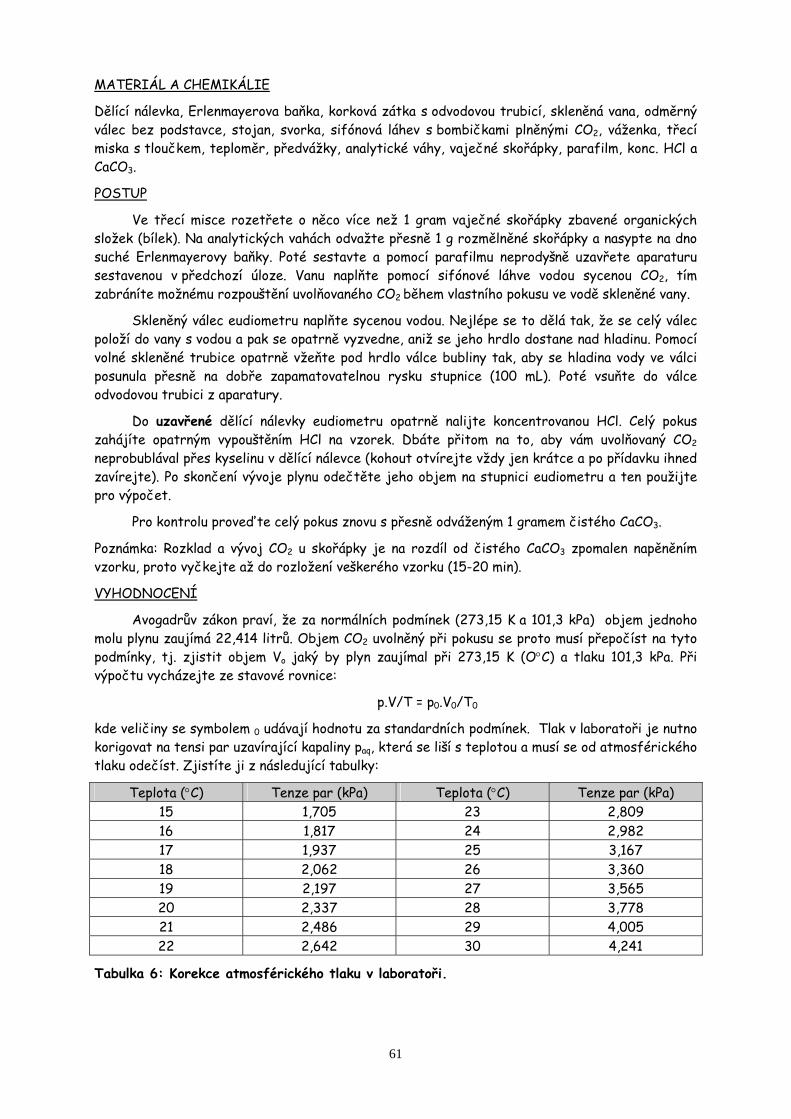
MATERIÁL A CHEMIKÁLIE
Dělící nálevka, Erlenmayerova baňka, korková zátka s odvodovou trubicí, skleněná vana, odměrný válec bez podstavce, stojan, svorka, sifónová láhev s bombičkami plněnými CO2, váženka, třecí miska s tloučkem, teploměr, předvážky, analytické váhy, vaječné skořápky, parafilm, konc. HCl a CaCO3.
POSTUP
Ve třecí misce rozetřete o něco více než 1 gram vaječné skořápky zbavené organických složek (bílek). Na analytických vahách odvažte přesně 1 g rozmělněné skořápky a nasypte na dno suché Erlenmayerovy baňky. Poté sestavte a pomocí parafilmu neprodyšně uzavřete aparaturu sestavenou v předchozí úloze. Vanu naplňte pomocí sifónové láhve vodou sycenou CO2, tím zabráníte možnému rozpouštění uvolňovaného CO2 během vlastního pokusu ve vodě skleněné vany.
Skleněný válec eudiometru naplňte sycenou vodou. Nejlépe se to dělá tak, že se celý válec položí do vany s vodou a pak se opatrně vyzvedne, aniž se jeho hrdlo dostane nad hladinu. Pomocí volné skleněné trubice opatrně vžeňte pod hrdlo válce bubliny tak, aby se hladina vody ve válci posunula přesně na dobře zapamatovatelnou rysku stupnice (100 mL). Poté vsuňte do válce odvodovou trubici z aparatury.
Do uzavřené dělící nálevky eudiometru opatrně nalijte koncentrovanou HCl. Celý pokus zahájíte opatrným vypouštěním HCl na vzorek. Dbáte přitom na to, aby vám uvolňovaný CO2 neprobublával přes kyselinu v dělící nálevce (kohout otvírejte vždy jen krátce a po přídavku ihned zavírejte). Po skončení vývoje plynu odečtěte jeho objem na stupnici eudiometru a ten použijte pro výpočet.
Pro kontrolu proveďte celý pokus znovu s přesně odváženým 1 gramem čistého CaCO3.
Poznámka: Rozklad a vývoj CO2 u skořápky je na rozdíl od čistého CaCO3 zpomalen napěněním vzorku, proto vyčkejte až do rozložení veškerého vzorku (15-20 min).
VYHODNOCENÍ
Avogadrův zákon praví, že za normálních podmínek (273,15 K a 101,3 kPa) objem jednoho molu plynu zaujímá 22,414 litrů. Objem CO2 uvolněný při pokusu se proto musí přepočíst na tyto podmínky, tj. zjistit objem Vo jaký by plyn zaujímal při 273,15 K (O°C) a tlaku 101,3 kPa. Při výpočtu vycházejte ze stavové rovnice:
p.V/T = p0.V0/T0
kde veličiny se symbolem 0 udávají hodnotu za standardních podmínek. Tlak v laboratoři je nutno korigovat na tensi par uzavírající kapaliny paq, která se liší s teplotou a musí se od atmosférického tlaku odečíst. Zjistíte ji z následující tabulky:
Teplota (°C) Tenze par (kPa) Teplota (°C) Tenze par (kPa) 15 1,705 23 2,809 16 1,817 24 2,982 17 1,937 25 3,167 18 2,062 26 3,360 19 2,197 27 3,565 20 2,337 28 3,778 21 2,486 29 4,005 22 2,642 30 4,241
Tabulka 6: Korekce atmosférického tlaku v laboratoři.
61

Po úpravě stavové rovnice dostáváte:
V0 = (p – paq).V.T0 / p0.T
Nezapomeňte, že teplotu T i T0 je nutno uvádět v Kelvinech, T získáte připočtením 273,15 k teplotě ve stupních Celsia. Současný atmosférický tlak v laboratoři vám sdělí vedoucí cvičení.
K výpočtu množství původního CaCO3 je pak nutné si uvědomit, že reakce probíhá stechiometricky podle koeficientů (z jednoho molu CaCO3 se uvolní jeden mol CO2), a dále že jeden mol je látkové množství soustavy, která obsahuje právě tolik částic (atomů, molekul, iontů či jiných částic), kolik je atomů v 0,012 kg uhlíku C12. Jeho hmotnost je číselně rovna poměrné molekulové (atomové, iontové) hmotnosti uvažované látky.
Určete procentuální obsah CaCO3 ve vaječné skořápce a správnost výpočtu i měření si ověřte výpočtem hmotnosti CaCO3, přes CO2 uvolněný jeho rozkladem z přesně naváženého 1 gramu CaCO3. Získané hodnoty komentujte a vyhodnoťte přesnost vašeho pokusu.
Doporučená literatura:
Eysseltová, J. a kol.: Základy laboratorní techniky, Nakladatelství Karolinium Praha, 2000. Kašpárek, F. a kol.: Cvičení z laboratorní techniky, Vydavatelství UP Olomouc, 2000. Pazdera, P. a Potáček, M.: Laboratorní cvičení z organické chemie, Vydavatelství UJEP Brno, 1984.
62

Laboratorní cvičení č. 8
SUŠENÍ, CHLAZENÍ A LYOFILIZACE
Sušením se rozumí proces, kdy zbavujeme látku pevnou, kapalnou či plynnou vody nebo jiné
nežádoucí kapaliny a jejích par. V praxi se nejčastěji setkáváme s odstraňováním vody, ve které většina chemických reakcí probíhá. A nebo naopak je třeba vodu odstranit z reakční směsi u reakcí, kde i v malém množství může průběh reakce nepříznivě ovlivnit. Vlhkost a vodu lze odstraňovat jednak fyzikálně – sušením v proudu horkého vzduchu, zahříváním nebo chemicky - vhodným dehydratačním činidlem. Podle způsobu jak vážou vodu lze tato činidla rozdělit do následujících skupin:
- látky hygroskopické, které snadno vytvářejí hydráty, patří sem bezvodé soli a jejich nižší hydráty (např. K2CO3, Na2SO4, KOH)
- látky odstraňující vodu ze směsi tím, že s ní chemicky reagují (P4O10, BaO, CaO, Na, B2O5)
- látky poutající vodu fyzikální adsorpcí (silikagel, Al2O3, některé polymery)
SUŠENÍ PLYNŮ
Plyny sušíme v promývačkách (obr. 27), sušících věžích (obr. 28) a U-trubicích (obr. 29), do kterých umísťujeme sušící činidlo - pevnou látku (do sušících věží a U-trubic) a hygroskopické kapaliny (do promývaček). Stupeň vysušení je závislý na tloušťce vrstvy sušícího činidla a dobrém styku plynu s ním. Toho můžeme dosáhnout vložením frity do promývačky a propojením několika promývaček za sebou. Sušící věže a U-trubice plníme tak, aby hmota kladla plynu všude stejný odpor a plyn případně neprocházel kanálky v ní. Promývačky plníme do 1/3 jejich objemu.
Obrázek 27: Promývačky. Obrázek 28: Sušící věž.
Při výběru sušidla je nutno brát v úvahu, zda je plyn inertní nebo zda má bazický či kyselý charakter. Bazické plyny, jako např. amoniak, nejčastěji sušíme pevnými hydroxidy. Kyselé (H2S, HBr) a inertní (N2) pak oxidem fosforečným, silikagelem či nejúčinněji kyselinou sírovou (tab. 7) Velmi účinným způsobem sušení plynu je jeho vymrazení. Provádí se tak, že plyn vedeme U-trubicí ponořenou do chladící směsi (viz níže).
63
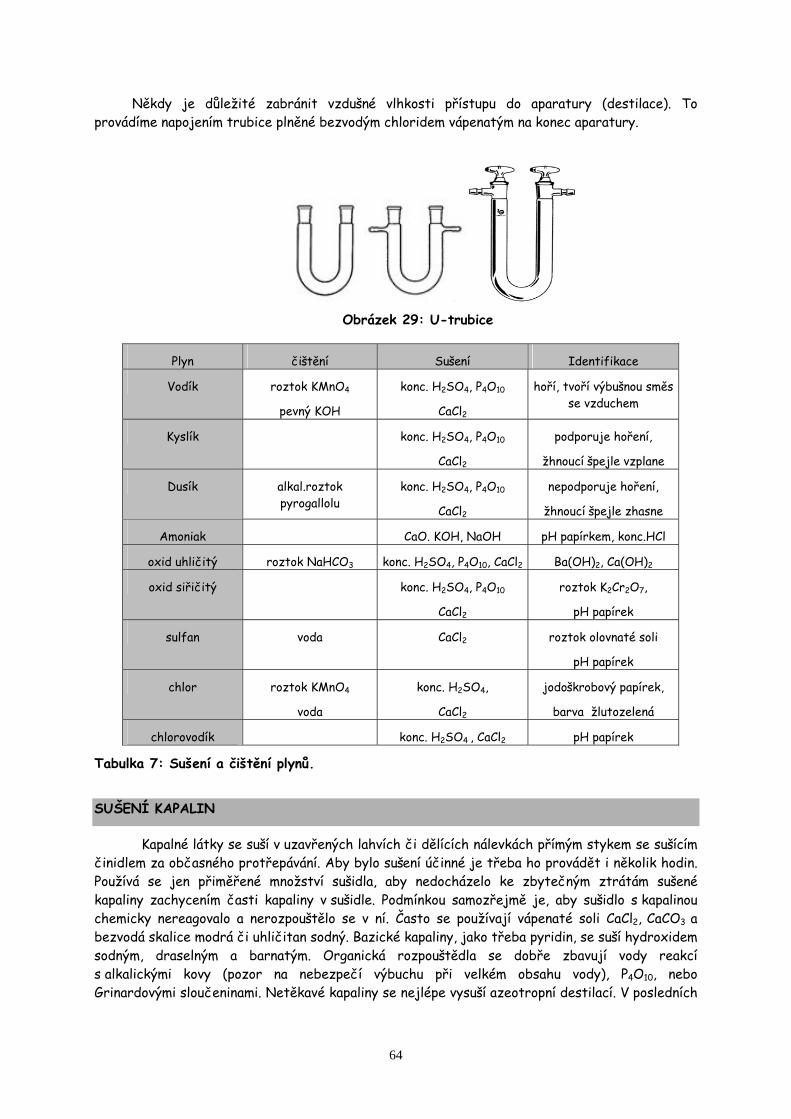
Někdy je důležité zabránit vzdušné vlhkosti přístupu do aparatury (destilace). To provádíme napojením trubice plněné bezvodým chloridem vápenatým na konec aparatury.
Obrázek 29: U-trubice
Plyn čištění Sušení Identifikace
Vodík roztok KMnO4
pevný KOH
konc. H2SO4, P4O10
CaCl2
hoří, tvoří výbušnou směs se vzduchem
Kyslík konc. H2SO4, P4O10
CaCl2
podporuje hoření,
žhnoucí špejle vzplane
Dusík alkal.roztok pyrogallolu
konc. H2SO4, P4O10
CaCl2
nepodporuje hoření,
žhnoucí špejle zhasne
Amoniak CaO. KOH, NaOH pH papírkem, konc.HCl
oxid uhličitý roztok NaHCO3 konc. H2SO4, P4O10, CaCl2 Ba(OH)2, Ca(OH)2
oxid siřičitý konc. H2SO4, P4O10
CaCl2
roztok K2Cr2O7,
pH papírek
sulfan voda CaCl2 roztok olovnaté soli
pH papírek
chlor roztok KMnO4
voda
konc. H2SO4,
CaCl2
jodoškrobový papírek,
barva žlutozelená
chlorovodík konc. H2SO4 , CaCl2 pH papírek
Tabulka 7: Sušení a čištění plynů.
SUŠENÍ KAPALIN
Kapalné látky se suší v uzavřených lahvích či dělících nálevkách přímým stykem se sušícím činidlem za občasného protřepávání. Aby bylo sušení účinné je třeba ho provádět i několik hodin. Používá se jen přiměřené množství sušidla, aby nedocházelo ke zbytečným ztrátám sušené kapaliny zachycením časti kapaliny v sušidle. Podmínkou samozřejmě je, aby sušidlo s kapalinou chemicky nereagovalo a nerozpouštělo se v ní. Často se používají vápenaté soli CaCl2, CaCO3 a bezvodá skalice modrá či uhličitan sodný. Bazické kapaliny, jako třeba pyridin, se suší hydroxidem sodným, draselným a barnatým. Organická rozpouštědla se dobře zbavují vody reakcí s alkalickými kovy (pozor na nebezpečí výbuchu při velkém obsahu vody), P4O10, nebo Grinardovými sloučeninami. Netěkavé kapaliny se nejlépe vysuší azeotropní destilací. V posledních
64

letech se s oblibou začaly na vysoušení organických rozpouštědel používat syntetické křemičitany.
SUŠENÍ PEVNÝCH LÁTEK
Ve velké většině případů je sušení pevných látek založeno na volném odpařování vlhkosti. To může probíhat za normální nebo zvýšené teploty, ve vakuu nebo za normálního tlaku. Před počátkem sušení je třeba vždy odstranit co největší množství vody mechanicky – dokonalým odsátím, vylisováním apod. Nejjednodušším způsobem sušení, je rozprostření sušené látky v tenké vrstvě na filtračním papíru či porcelánové misce na volném vzduchu. Je možné provést sušení za zvýšené teploty v sušárně nebo pod infralampou. Filtrační papír však snáší teploty jen do 100ºC, při vyšších teplotách je třeba použít vhodnější podložku. V sušárně nikdy nesmějí být sušené látky těkavé nebo látky se zbytky těkavých hořlavých rozpouštědel, jejichž páry by mohly se vzduchem vytvořit třaskavou směs. Pro sušení při teplotách vyšších než 200ºC se používají muflové pece nebo sušící pícky.
Obrázek 30: Exsikátory: klasický vakuový
Chemickou cestou je možno vysoušet různými dehydratačními činidly. Vysoušení obvykle provádíme v uzavřených nádobách – exsikátorech, které mohou být i pod vakuem (obr. 30). Rychlejšího vysoušení malých množství látek lze dosáhnout v tzv. vysoušecích pistolích (Abderhaldenův přístroj) rovněž napojených na vakuum (obr. 31). Látka (A) je zde vyhřívána párami vroucí kapaliny o vhodném bodu varu (C), kdežto sušidlo je ve zvláštní baňce mimo vyhřívaný prostor (B). Nejběžnější vysoušecí prostředky používané v exsikátorech jsou shrnuty v tabulce 8. Kyselinu sírovou a oxid fosforečný nenaléváme přímo na dno exsikátoru, ale zvlhčíme kapalinou skleněné kuličky a tím zvětšíme absorpční povrch.
BC
A
Obrázek 31: Abderhaldenův přístroj.
65
Sušidlo zbylé mg H2O na litr sušeného vzduchu při 25ºC bezv. CuSO4 1,4 98% H2SO4 0,3 bezv. CaCl2 0,25 – 0,14 CaO 0,2 Mg(ClO4)2.3H2O 0,031 silikagel 0,02 100% H2SO4 0,003 KOH 0,002 bezv. Mg(ClO4)2 0,0005 P4O10 0,000025
Tabulka 8: Sušidla vhodná pro použití v exsikátorech a jejich účinnost.
CHLAZENÍ
V chemické laboratoři se velmi často setkáváme s chlazením. Chlazení hraje důležitou roli při krystalizačních pochodech, při destilaci látek s nízkou teplotou bodu varu. Chladit musíme silně exotermické reakce.
Při izolacích biomolekul, u nichž je nutné zachovat jejich biologickou funkci (enzymová aktivita) je třeba celý proces izolace provádět při teplotě okolo 0ºC. Tím zabraňujeme denaturaci proteinů, která in vitro nastává již při teplotách nad 4ºC. Ve velkých biochemických ústavech k tomuto účelu slouží celé speciální chladící místnosti, tzv. „cold roomy“. V našich podmínkách můžeme chlazení při izolaci dosáhnout použitím misek s drceným ledem, popř. provádět nenáročné úkony (dialýza) v ledničkách (viz. lab. cvičení č. 3).
K dlouhodobému uchovávání mnohých chemikálií a biologického materiálu je třeba snížené teploty. V biochemické laboratoři proto velice často narazíme na lednice a mrazící boxy vyhrazené k tomu účelu. Pro uchovávání vzorku RNA a kompetentních bakteriálních řetězců je pak nezbytný hlubokomrazící box s teplotou -80ºC. Případně se mohou tyto vzorky uchovávat v Dewarových nádobách s tekutým dusíkem (-196ºC, obr. 32). Tekutý dusík se také používá na rozrušování a snadnější homogenizaci biologického materiálu. Dewarovy nádoby mají dvojité stěny mezi nimiž je vakuum. Stěny bývají většinou postříbřeny, aby docházelo k menším ztrátám sáláním. Chlazení kapalným vzduchem se moc často nepoužívá (-180ºC), jelikož při manipulaci s ním hrozí nebezpečí exploze.
Obrázek 32: Dewarova nádoba. Průřez a celkový pohled.
Nejčastěji se však setkáváme s chlazením vodou. Běžná vodovodní voda mívá teplotu kolem 12ºC a pomocí různých průtokových systémů (chladič) můžeme dosáhnout jejího rovnoměrného proudění kolem chlazených nádob. Účinnějšího chlazení než je chlazení drceným ledem, lze zajistit chladícími směsmi, což jsou směsi drceného ledu s elektrolytem. Při přípravě směsi je třeba dbát na dobré promísení obou složek a na to, aby byl zabezpečen dokonalý styk chlazeného povrchu s chladícím prostředím. Směsi NaCl s ledem (1:3) lze dosáhnout teploty -21ºC. Nejúčinnější je pak směs CaCl2.6H2O (3:2) s teplotou -49ºC. Dalším poměrně snadno dostupným
66

chladícím prostředkem je pevný oxid uhličitý – suchý led. Používá se ho nejčastěji ve směsi s různými nízkotuhnoucími organickými rozpouštědly, ve kterých se rozpouští a která ochlazuje často na teploty nižší, než je jeho bod sublimace. Unáší-li plynný oxid uhličitý s sebou páry těkavého rozpouštědla směs se dále ochlazuje v důsledku odpařování kapaliny. Použitím následujících rozpouštědel s pevným CO2 docílíme těchto teplot:
ethylenglykol -15ºC chloroform -77ºC aceton -86ºC ether -90ºC absolutní ethanol -100ºC
LYOFILIZACE – MRAZOVÁ SUBLIMACE
Lyofilizace je jedním z nejšetrnějších způsobu odstraňování vody z vodných roztoků. Používá se především k zakoncentrovávání roztoků biologicky aktivních látek, které jsou náchylné na vyšší teploty a nemohou být zakoncentrovány klasickými způsoby (destilace, odpařování na vakuové rotační odparce). Podstatou lyofilizace je odsublimování vody ze zamraženého vzorku pod vakuem. Vakuum by mělo být silné okolo 1-3 kPa. Lyofilizovat se dají pouze vodné roztoky látek. Přítomnost organických rozpouštědel je nežádoucí, protože snižuji teplotu tuhnutí vody a zvyšují riziko roztopení vzorku během lyofilizace. Nedostatečné zmrazení vzorku a jeho roztopení během lyofilizace se projevuje napěněním a může způsobit denaturaci nativního proteinu (ztráta enzymové aktivity). Vzorek před lyofilizací je nutno odsolit, jelikož se po jeho zakoncentrování (odstranění většího podílu vody) zvedne koncentrace soli nad mez rozpustnosti. Zvýšená pozornost by se měla také věnovat lyofilizaci tlumivých roztoků. Rozdílná krystalizace solí ve vícesložkových pufrech může zapříčinit značné posuny hodnoty pH a způsobit tak opět denaturaci proteinu.
Na lyofilizaci větších objemů se používají skleněné nádoby, které vydrží zamrazení na 70°C. Menší objemy lze zamrazovat a lyofilizovat přímo v plastových mikrozkumavkách. Ústí nádoby, ve které se nachází vzorek, je třeba při lyofilizaci zakrýt porézním materiálem, jelikož lyofilizovaný produkt je velmi lehký a prachovitý a mohl by ho proud vodní páry strhnout do kondenzátoru. Vzorky se předem vychlazují buď několikahodinovým zamrazením v hlubokomrazících boxech (-80°C), nebo umístěním do chladící lázně zhotovené z pevného CO2 a acetonu. Mikrozkumavky se vzorkem mohou být zamraženy přímo krátkým ponořením do kapalného dusíku. Před začátkem vlastní lyofilizace je třeba prostor pro kondenzaci vodních par dokonale vychladit. Před koncem lyofilizace se do systému pomalu vpouští vzduch a po zrušení vakua se odeberou vzorky a vypne vývěva. Kdyby se vývěva vypnula před zrušením vakua, mohl by se olej z vývěvy nasát do kondenzačního systému.
Vlastní zařízení na lyofilizaci (obr. 33) se liší typ od typu podle výrobce. Obecně se však vždy skládá ze zdroje vakua - výkonné olejové vývěvy, mrazící jednotky spojené s prostorem pro uložení vzorku a kondenzací par. Přístroj je pak vždy opatřen kontrolním a regulačním zařízením pro registraci parametrů (teplota, tlak) během lyofilizace. Některé moderní lyofilizátory bývají vybaveny navíc chlazenou centrifugou, do které se umísťují lyofilizované vzorky. Odstředivou silou je pak zabráněno vybublání či vypěnění vzorku, který se před vlastní lyofilizací již nemusí zamrazovat a tím se celý proces značně urychluje.
67

Obrázek 33: Základní princip jednoduchého lyofilizátoru.
V laboratořích, které nedisponují finančně nákladným zařízením na lyofilizaci, lze tepelně nestabilní vodné roztoky biopolymerů zakoncentrovávat ultrafiltrací (viz. lab. cvičení č. 3) nebo ještě jednodušším způsobem založeném na principu gelové filtrace. Do roztoku se přidá asi třetina jeho objemu suchého hydrofilního gelu, např. Sephadex G-25, používaného pro molekulovou vylučovací chromatografii (gelovou filtraci, viz. lab. cvičení č. 11). Voda a nízkomolekulární látky se pohlcují částečkami Sephadexu, který bobtná a vysokomolekulární látky zůstávají v roztoku. Po několika minutách se Sephadex oddělí od roztoku centrifugací. Proces se dá samozřejmě i několikrát opakovat. Nevýhodou jsou však značné ztráty biopolymeru. Během zakoncentrovávání nedochází ke změnám pH ani iontové síly.
ÚKOL č. 14: Stanovení obsahu krystalové vody ve skalici modré
U mnohých krystalických látek se účastní stavby jejich struktury i molekuly vody. Takové krystaly se nazývají hydráty a vázaná voda vodou krystalovou. Jako příklad lze uvést dihydrát chloridu barnatého BaCl2.2H2O, skalici zelenou FeSO4.7H2O aj. Kromě látek tvořících pouze jediný hydrát, jsou známy i takové, které tvoří hydrátů několik. Obsah krystalové vody potom závisí na teplotě, při které látka krystalizuje. Uhličitan sodný vyloučený z roztoku za varu je monohydrát, krystalický za laboratorní teploty je dekahydrát (obsahuje deset molekul vody) a jeho preparát vykrystalovaný při -20ºC obsahuje patnáct molekul vody, tzv. pentadekahydrát. Hydráty krystalizující při nižší teplotě mívají zpravidla vyšší obsah hydrátové vody. Obsah hydrátové vody bývá často ovlivněn jinou složkou v roztoku. Např. přítomnost kyseliny snižuje stupeň hydratace. Tak síran měďnatý může z roztoku obsahující nadbytek kyseliny sírové krystalizovat i při laboratorní teplotě jako tri- nebo monohydrát.
Stálost hydrátů závisí na parciálním tlaku vodní páry nad příslušným hydrátem. Je-li tento parciální tlak stejný jako tense vodních par v okolí, je hydrát na vzduchu stálý. Je-li tento parciální tlak větší, než je tense vodní páry v okolí, ztrácí krystal samovolně část hydrátové vody, aby se rozdíl tenzí vyrovnal. V opačném případě, je-li tense vodních par v okolí vyšší, hydrát
68

samovolně přijímá vodu z okolí a dochází k jeho vlhnutí případně až rozpouštění. Látky, které ochotně přijímají vodu z prostředí, se označují jako hygroskopické.
Hydrát lze částečně nebo úplně zbavit krystalové vody tím, že se tense jeho vodních par zvýší zahřáním nebo že se značně sníží tense vodních par v okolí (tj. umístěním krystalohydrátu do exsikátoru nad silně hygroskopickou látku. Pro dehydrataci zahřáním stačí obvykle teplota mezi 100-300ºC. Při použití teploty vyšší může kromě dehydratace docházet i k hlubšímu rozkladu látky. Před dehydratací je třeba látku vždy důkladně rozetřít. Neučiní-li se tak, může unikající vodní pára větší krystaly roztrhat a dochází ke ztrátám vyletováním drobných částic a prskání z kelímku.
MATERIÁL
Elektrická sušárna, třecí miska s tloučkem, porcelánový kelímek, analytické váhy, kleště, exsikátor, lžička, pentahydrát síranu měďnatého.
POSTUP
V sušárně s teplotou vyregulovanou na 150ºC vysušte porcelánový kelímek do konstantní hmotnosti. Tzn. že se hmotnost kelímku po dalším sušení již nesnižuje. Hmotnost kelímku stanovte asi po 1/2 hodině sušení a následně ověřte vždy po 10 minutách. Hmotnost vysušeného kelímku si zapište. Manipulaci s kelímkem provádějte v exsikátoru, kde ho taky necháte po sušení vychladnout. Kelímku se nedotýkejte rukou, ale vždy kleštěmi. Rukou přenášíte na kelímek zpět vlhkost. Mezitím rozetřete asi 1,5 gramu skalice modré ve třecí misce. A do vysušeného a zváženého kelímku přesně odvažte asi 1 g rozetřené CuSO4.5H2O (tzn. na analytických vahách stanovíte hmotnost přibližně 1 gramu s přesností na 4 desetinná místa, např. budete mít 1,0584 g skalice modré). Sušárnu vyregulujte na 110ºC a kelímek s pentahydrátem v ní sušte opět do konstantní hmotnosti (asi 1/2 hodiny). Po vychladnutí v exsikátoru přesně zvažte.
VYHODNOCENÍ
Podle úbytku hmotnosti určete, kolik molekul vody ztratil pentahydrát síranu měďnatého při teplotě 110ºC.
ÚKOL č. 15: Příprava bezvodého silikagelu
Silikagel je amorfní forma oxidu křemičitého. Je netoxický a chemicky odolný vůči většině kyselin, je však citlivý vůči zásaditým látkám. Silikagel je používán především ve formě granulátu a kuliček, což značně zvětšuje jeho povrch a umožňuje tak, aby lépe vázal vzdušnou vlhkost. Správně připravený silikagel může vázat vlhkost až do 40% své váhy. K indikaci nasycenosti silikagelu se používá chlorid kobaltnatý, do jehož roztoku se silikagel vměšuje, a tím získává typickou narůžovělou barvu. Po vysušení CoCl2 ztrácí krystalickou vodu a mění zabarvení na intenzivní modř. Modrý silikagel je tedy připraven k použití. Adsorpcí vody pak postupně znovu přechází do růžového zabarvení a je opět nutno ho regenerovat vysušením.
MATERIÁL
Elektrická sušárna, kleště, exsikátor, lžička, kádinka, porcelánová miska, hexahydrát chloridu kobaltnatého, technický silikagel.
POSTUP
Do 100 mL kádinky navažte 20 g technického silikagelu. Do další kádinky si připravte 10 mL přibližně 4% roztoku CoCl2.6H2O. A tento roztok vlijte do silikagelu a pořádně promíchejte. Narůžovělý preparát vysušte mezi filtračními papíry a rozestřete na porcelánovou misku v co nejtenčí vrstvě. Misku vložte do sušárny a vyregulujte teplotu na 150ºC. Přibližně každých 15
69

minut silikagel v misce promíchejte skleněnou tyčinkou a sušte až do doby, kdy má intenzivně modrou barvu (asi 1 hod). Modrým silikagelem naplňte dno exsikátoru nebo ho uložte do suché zábrusové láhve (rozhodne vedoucí cvičení).
ÚKOL č. 16: Příprava, sušení, čištění a důkaz vodíku
Vodík je nejlehčí plyn, bez chuti, bez zápachu. Jeho teplota varu se pohybuje okolo -252,7ºC stupňů Celsia a teplota tání je -259ºC. Ve vodě se rozpouští velmi málo a téměř vždy (kromě stavu zrodu) se vyskytuje ve formě dvouatomových molekul. Vodík jako plyn je směsí svých tří izotopů. Prócium - má v jádře pouze jeden proton, deuterium neboli těžký vodík má ve svém jádře kromě protonu také jeden neutron, a proto má i vlastní značku - D, tricium - jeho jádro obsahuje již dva neutrony. Zjistilo se však, že existují i izotopy s nukleonovým číslem 4 a 5. Ve sloučeninách s vodíkem bývá vazba polární. Vodík má ve sloučeninách většinou kladný náboj, ale s některými kovy se váže ve formě aniontu a vznikají tzv. hydridy. S přechodnými kovy tvoří sloučeniny, které mají charakter tuhých roztoků a tvoří v nich také někdy kovovou vazbu. Celkově se slučuje více s nekovovými než kovovými prvky. Za normální teploty je málo reaktivní, ale pokud se teplota zvýší, slučuje se dobře s kyslíkem a halovými prvky za uvolnění velké tepelné energie. Tento prvek má silně redukční vlastnosti - za zvýšené teploty redukuje např. oxidy, sulfidy nebo chloridy až na elementární kov. Vodík se dá získat uvolněním z vody alkalickými kovy nebo kovy alkalických zemin již za studena (většina ostatních kovů ho uvolňuje z vody až za zvýšené teploty). Uvolňuje ho i rozžhavený uhlík. Nejčastěji se však připravuje buď elektrolýzou vody nebo reakcí zinku s kyselinou chlorovodíkovou případně sírovou.
Pozor: Při práci s vodíkem je třeba dbát zvýšené opatrnosti a dodržovat bezpečnostní předpisy, hrozí nebezpečí vzniku třaskavé směsi se vzduchem.
MATERIÁL
Dělící nálevka, frakční baňka, korková zátka, skleněné trubice, hadičky, U-trubice, promývačka, vana, špejle, kyselina chlorovodíková, práškový zinek, hydroxid draselný, konc. kyselina sírová, dětský bublifuk.
POSTUP
Sestavte aparaturu pro přípravu vodíku sestávající z dělící nálevky a frakční baňky. Korkovou zátkou ve frakční baňce vyveďte skleněnou trubici, na kterou postupně napojíte U-trubici naplněnou pevným KOH a promývačku s konc. kyselinou sírovou. U-trubici a dělící nálevku řádně upevněte do stojanu. Na vývod z promývačky napojte skleněnou trubici asi o délce 20 cm. Připravte si skleněnou vanu naplněnou do 1/4 vodou. Na dno frakční baňky nasypte přáškový zinek a do dělicí nálevky nalijte přibližně 20 mL kyseliny chlorovodíkové (ředění vodou 1:1). Zkontrolujte, zda jsou všechny spoje řádně napojeny, a zaveďte skleněnou trubici z promývačky pod hladinu vody ve vaně.
Opatrně začněte přikapávat HCl na zinek a sledujte vývoj vodíku (unikající bubliny do vany). Skleněnou trubicí ve vaně vyjměte a snažte se vyfukovat bubliny bublifukem. Pokud vývoj vodíku ustane přilijte na zinek další podíl kyseliny chlorovodíkové. Když máte vodík vznikající v aparatuře dostatečně vyčištěný, budou vám vznikající bubliny létat vzhůru ke stropu. Hořící špejlí opatrně bubliny zapalujte.
Nikdy nedávejte zapálenou špejli k ústí přívodové trubičky z aparatury na přípravu vodíku. Nebezpečí výbuchu!!!
70

VYHODNOCENÍ
1) Popište co se stalo, když jste přiblížili špejli k bublinám.
2) Proč jste na čištění a sušení vodíku použili H2SO4 a KOH? K jakým chemickým dějům dochází?
ÚKOL č. 17: Zakoncentrování roztoku proteinů pomocí gelové filtrace
V biochemické laboratoři se budete velice často setkávat s požadavkem na zakoncentrovávání naředěných roztoků biopolymerů. Existuje řada způsobů, jak snížit v roztoku množství vody a nízkomolekulárních látek tak, aby vysokomolekulární látka zůstala ve stejném množství. Lze k tomu využít dialýzy, ultrafiltrace či lyofilizace. Tyto procesy jsou však většinou dosti zdlouhavé a musí se řešit problém denaturace biologicky aktivních biopolymerů. Jeden z nejrychlejších a nejšetrnějších způsobu zahuštění vysokomolekulární látky je založen na principu gelové filtrace a prakticky ho provedete se vzorkem standartního hovězího sérového albuminu (BSA – bovine serum albumin).
MATERIÁL
Automatické pipety, Hamiltonova pipeta, mikrozkumavky, zkumavky, špachtle, vortex, stolní pikofuga, spektrofotometr, kyveta, kalibrační křivka pro metodu Bradfordové, roztok BSA, suchý Sephadex G-50 Coarse, Bradfordové činidlo.
POSTUP
Do mikrozkumavky odpipetujte přesně 1 mL ze zásobní láhve roztoku BSA a přisypte špachtlí suchý Sephadex G-50 tak, aby po nabobtnání zabral asi 1/3 objemu (na špičku špachtle). Směs nechte asi 10 minut stát za laboratorní teploty a za občasného promíchání na vortexu. Poté mikrozkumavku asi 2 minuty centrifugujte na pikofuze. Do čisté mikrozkumavky opatrně přeneste čirý supernatant nad usazeným Sephadexem a pomocí automatické pipety nejpřesněji odhadněte jeho objem. Objem si zaznamenejte a do čisté mikrozkumavky odeberte 12 µL a označte jako vzorek 2. Ke zbylému supernatantu opět přisypte suchý Sephadex asi do 1/3 objemu, za občasného vortexování opět nechejte stát 10 minut a po 2 minutové centrifugaci odeberte a přesně změřte pipetou objem supernatantu. 12 µL odeberte do čisté mikrozkumavky jako vzorek 3. Se zbytkem celý proces ještě jednou zopakujte a dostanete tak vzorek 4.
Metodou Bradfordové změřte obsah proteinů vždy v 5 µL vzorku. Každé měření provádějte 2x. Nachystejte si 9 zkumavek do každé pipetujte 2 mL Bradfordové činidla a pomocí Hamiltonové pipety přidejte 5 µL vzorku, 9 zkumavka je blank (slepý vzorek, pro vynulování spektrofotometru). Vzorek číslo 1 je zásobní roztok BSA. Zkumavky s činidlem a vzorkem krátce zvortexujte a nechejte 5 minut stát při laboratorní teplotě. Poté změřte absorbanci při 595 nm. Hodnoty si zaznamenejte a pomocí kalibrační křivky stanovte obsah proteinu.
VYHODNOCENÍ
Sestavte si tabulku z naměřených hodnot – objem, obsah proteinu v 1 µL, celkový obsah proteinu ve vzorku. Vypočtěte kolikrát se vám podařilo zahustit původní roztok BSA a jak velkých ztrát jste se přitom dopustili (tzn. kolik BSA vám zbylo z původního množství obsaženého v 1 mL zásobního roztoku).
71

Doporučená literatura:
Ferenčík, M. a kol.: Biochemické laboratórne metódy, Alfa Bratislava, 1981. Kašpárek, F. a kol.: Cvičení z laboratorní techniky, Vydavatelství UP Olomouc, 2000. Mareček, A. a Růžička, A.: Laboratorní technika pro nechemické obory, Vydavatelství MU Brno, 1997. Klečková, M. a Šindelář, Z.: Školní pokusy z anorganické a organické chemie, Vydavatelství UP Olomouc, 1993.
72

Laboratorní cvičení č. 9
KRYSTALIZACE, PROMÝVÁNÍ NA FILTRU, DEKANTACE A STANOVENÍ BODU TÁNÍ
KRYSTALIZACE
Krystalizace je proces vylučování pevné fáze z taveniny nebo rozpuštěné pevné látky z roztoku. Z našeho hlediska má význam pouze krystalizace z roztoku, což je jedna ze základních metod čištění pevných látek.
Čištění látek krystalizací spočívá v jejím rozpuštění ve vhodném rozpouštědle, vyčištění roztoku filtrací a opětné vyloučení látky z roztoku v krystalické formě. Látku lze ke krystalizaci přivést několikerým způsobem:
− Krystalizace volným odpařováním. Volným odpařováním roztoku za normální teploty dojdeme ke stavu, kdy je roztok při dané teplotě nasycen a látka se pak vylučuje z roztoku ve velkých, dobře vyvinutých krystalech.
− Krystalizace volným chladnutím. Při tomto způsobu krystalizace si nejprve připravíme za varu nasycený roztok, přefiltrujeme a kádinku s vroucím roztokem vložíme do termostatu, který udrží roztok několik hodin teplý. Krystalky, které po vychladnutí roztoku získáme, jsou velké a pěkně vyvinuté.
Výše popsané dva způsoby krystalizace jsou časově náročné a hodí se především pro čištění látek, jejichž rozpustnost není výrazně závislá na teplotě.
− Rušená krystalizace. Opět nejdříve připravíme za horka (nebo varu) nasycený roztok a ten pak za trvalého míchání krouživým pohybem krystalizační nádoby prudce ochladíme na teplotu tekoucí vody. Získáme tak velké množství drobných krystalů, které odfiltrujeme od tzv. matečného louhu, jenž obsahuje zbytek krystalizované látky a většinu nečistot. Novým odpařením matečného louhu můžeme získat další, ovšem již méně čisté podíly. Rušené krystalizace užíváme u látek, u nichž je rozdíl v rozpustnosti za studena a za horka dostatečně velký.
− Krystalizace vyvolaná změnou složení rozpouštědla. Změnou složení rozpouštědla lze z roztoku vyloučit tu látku, která je v dané směsi nerozpustná. V praxi to provádíme tak, že k vodnému roztoku přidáváme alkohol, aceton nebo jiné méně polární rozpouštědlo, které je mísitelné s vodou. V takto získané směsi se brzy překročí součin rozpustnosti rozpouštěné látky, která se pak začne vylučovat v krystalech.
O nasycenosti roztoku se přesvědčíme u rychle krystalizujících látek např. tak, že kapku roztoku přeneseme tyčinkou na hodinové sklíčko. Když se roztok zakalí vyloučenými krystaly, je již dostatečně zahuštěn. U řady látek se na hladině lehkých nasycených roztoků vytváří vlivem chladného okolního vzduchu tzv. krystalizační blána (plovoucí krystaly).
Aby látka krystalizovala z roztoku, musí být porušená fázová rovnováha dané soustavy, tj. roztok musí být přesycen. Dále je pro krystalizaci nutné, aby vznikala nepatrná krystalizační centra, jež pak narůstají v krystaly. Vlastní pochod krystalizace se skládá z tvorby jader, k niž dojde porušením fázové rovnováhy roztoku ochlazením na teplotu nižší než je mez nasycenosti, a z růstu krystalů na vytvořených jádrech. Tvorba jader závisí na intenzitě chlazení, na rychlosti a způsobů míchání, na teplotě, na vlastnostech látky a také na nečistotách obsažených v roztoku. Je známo, že vznik velkého počtu jader je podporován rychlým ochlazením, intenzivním mícháním a malou molární hmotností rozpuštěné látky. Počet vznikajících jader se projevuje na tvaru a velikosti krystalů. Při malém počtu jader se pomalu tvoří velké krystaly s plně vyvinutými plochami. Velký počet malých krystalizačních center podmiňuje naopak vznik drobných krystalů

se špatně vyvinutými plochami. Všechny uvedené faktory pak mají vliv na krystalizační výtěžek, což je poměr množství vykrystalizované látky k výchozímu množství látky.
Jemné krystaly zadržují na svém povrchu mnoho matečného louhu, špatně se filtrují a promývají. U velkých krystalů hrozí naopak nebezpečí zadržení matečného louhu v eventuálních dutinách. Pro získání čisté látky je proto důležité i udržení přiměřené zrnitostí krystalů.
Některé látky mají sklon k tvorbě přesycených roztoků, to znamená, že ani po přestoupení meze rozpustnosti, např. ochlazením, látka nekrystalizuje. V takovém případě stačí někdy k vyvolání krystalizace tření stěny kádinky ostrohrannou skleněnou tyčinkou. Když i to selhává je možno vyvolat krystalizaci naočkováním, tj. vhozením několika krystalků látky do roztoku.
Doba trvání krystalizace je velmi různá a nelze ji všeobecně předepsat. V některých případech stačí pouze vyčkat na ochladnutí roztoku, jindy je nutné ponechat při nízké teplotě řadu dní. Výsledek krystalizace rovněž závisí na správné volbě rozpouštědla. Při jeho výběru je třeba brát v úvahu, že každá látka se nejlépe rozpouští v rozpouštědlech, která jsou ji chemicky nejpříbuznější. Důležitým požadavkem při volbě rozpouštědla je to, aby rozdíl rozpustnosti za horka a za studena byl co největší. V případě, že se takové rozpouštědlo nepodaří nalézt, je nutno část rozpouštědla odstranit odpařením. Přílišné odpaření rozpouštědla není však žádoucí, protože se tím čistící efekt krystalizace snižuje. Odpaření rozpouštědla lze provést několika způsoby:
− volným odpařením na vzduchu nebo v exsikátoru. Tento způsob se obyčejně používá jen pro rozpouštědla s nízkým bodem varu.
− odpařením na vodní lázní. Při postupném odpařování rozpouštědla postupně dochází k vylučování krystalů, které je nutno tyčinkou občas promíchat, aby došlo k porušení krystalické blány tvořící se na povrchu kapaliny.
− oddestilováním rozpouštědla za normálního nebo sníženého tlaku. Tento způsob je nejekonomičtější (regenerace rozpouštědla).
Na závěr je nutno připomenout fakt, že i když u většiny látek rozpustnost s teplotou roste, existuji též látky, jejichž rozpustnost je v určitém rozpouštědle prakticky nezávislá na teplotě (chlorid sodný ve vodě), případně i látky, jejichž rozpustnost s rostoucí teplotou klesá (např. monohydrát uhličitanu sodného ve vodě).
PROMÝVÁNÍ NA FILTRU A DEKANTACE
Promývání na filtru slouží k oddělování vyloučené sraženiny z roztoku od matečného louhu, nehledě na to, zda je sraženina amorfní či krystalické povahy. Tuto operaci lze provést několika způsoby: centrifugací, filtrací, odsátím nebo přímo dekantací v reagenční nádobě.
Při dekantaci necháme sraženinu usadit na dně nádoby, ve které ke srážení došlo a čirý matečný louh jednoduše odlijeme. Na dně nám kromě sraženiny samozřejmě zůstane ještě spousta matečného louhu i s nečistotami. Abychom tyto nečistoty odstranili, přilijeme čiré rozpouštědlo, dobře promícháme, sraženinu necháme opět usadit a čirý roztok dekantujeme. Tuto operaci můžeme opakovat několikrát, až do té doby než se zbavíme veškerých stop nečistot, o čemž se obvykle přesvědčíme příslušnou kvalitativní zkouškou. Dekantace se většinou používá na promývání sraženin amorfních, které se nedají promýt na filtru, jelikož ucpávají póry filtru. Dekantace se nehodí na promývání krystalů po krystalizaci. Při přídavku čirého rozpouštědla by samozřejmě docházelo ke zpětnému rozpouštění krystalů a tím k velkým ztrátám produktu.
Při promývání krystalů po krystalizaci je třeba řádně odkapat matečný louh a jeho zbytky odstranit promytím. Je nutné zvážit jakou kapalinu a jaké množství na promytí použít. Většinou se promytí provádí malým množstvím rozpouštědla, použitým pro krystalizaci. Při promývání je třeba dbát značné opatrnosti, zvláště tehdy, je-li produkt v rozpouštědle dobře rozpustný. V tomto případě může totiž dojít ke ztrátám produktu. Nejdokonalejší promytí lze dosáhnout, je-li
74

produkt na filtru při odpojené vývěvě promíchán s čistým rozpouštědlem, které je pak znova odsáto. Promývání je vhodné několikrát opakovat.
Matečný louh odsáváme na Büchnerově nálevce, do které vložíme papírový filtr o vhodném průměru. Papír by měl zakrýt všechny póry v nálevce, ale neměl by vystupovat nad její dno. Pokud je krystalizace prováděná v agresivním rozpouštědle, které by mohlo reagovat s celulosovým filtrem, je vhodnější použít na odsátí a následné promytí skleněnou fritu, do které se již filtrační papír nevkládá. Nevýhodou těchto frit je jejich časté ucpávání a špatné čištění. Po důkladném odsátí matečného louhu odpojíme vývěvu a malým množstvím čistého rozpouštědla, nejlépe ze střičky, omýváme krystaly. Rozpouštědlo smývá z povrchu krystalů zbylé stopy matečného louhu a případně rozpouští i zbytky nezreagovaných reakčních komponent. Po krátkém působení promývací kapalinu vždy odsajeme, toto opakujeme několikrát. Abychom zamezili větším ztrátám, je vhodné používat promývací kapalinu co nejstudenější. Při promývání je nutno mít na zřeteli, že je mnohem efektivnější promývat sraženinu vícekrát malými podíly promývacího roztoku než celým množstvím najednou. Je pravidlem neplnit Büchnerovu nálevku výše než pět milimetrů nad filtr.
Zmínku zasluhuje i správné oddělení pevné fáze od filtru. Jestliže ze skleněné frity můžeme produkt seškrabat tyčinkou nebo špachtlí, z Büchnerovy nálevky produkt v žádném případě nevyškrabujeme nebo nedáváme sušit i z nálevkou, protože tak bychom produkt znečistili částečkami filtračního papíru. Správný postup spočívá v přiklopení nálevky hodinovým sklem vhodného průměru, otočení stonkem vzhůru a silným fouknutím do stonku. Filtr s produktem vypadne na sklo, kde ho osušíme lehkým přitlačením čtverečku filtračního papíru. Produkt potom sloupneme z filtračního papíru.
ÚKOL č. 18: Izolace kyseliny citrónové z citrónové šťávy
Kyselina citronová - C3H4OH(COOH)3 je bílá krystalická látka rozpustná ve vodě a slabě rozpustná v organických rozpouštědlech. Její vodné roztoky jsou o něco kyselejší než kyselina octová. Stopy kyseliny citrónové lze nalézt v mnoha rostlinných i živočišných organismech, protože patří mezi intermediáty bazálního metabolismu (Krebsův cyklus). Velké množství této kyseliny je obsaženo ve šťávě citrusových plodů, ze které ji lze vysrážet vápenatými ionty. Ze vzniklého citrátu vápenatého je možné vlastní kyselinu citrónovou vytěsnit silnější kyselinou sírovou za vzniku síranu vápenatého. Komerčně se získává fermentací cukrů plísní Aspergillusniger. Používá se jako aditivum do jídel a nápojů pro vytvoření příjemné kyselé chuti. Dále se využívá v medicíně, textilnictví (modrotisk) a jako leštící prostředek na kovy.
MATERIÁL
Vařič, vodní lázeň, exsikátor, lis na citróny, kádinky, Pasteurovy pipety, skleněná tyčinka, pH papírky, Bűchnerova nálevka, odsávací baňka, vývěva, filtrační papír, skleněná frita se zábrusovou baňkou, porcelánová krystalizační miska, citrón popř. citrónová šťáva, 20% amoniak, 25% roztok CaCl2, 30% kyselina sírová.
POSTUP
Pomocí kuchyňského lisovače citrónů vymačkejte šťávu z jednoho citrónu a přelijte ji do 100 mL kádinky. 20% amoniakem šťávu zneutralizujte. Pasteurovou pipetou opatrně přidávejte za stálého míchání amoniak a pomocí pH papírku kontrolujte hodnotu pH. Ve slabě zásaditém prostředí šťáva tmavne. Silně zásadité prostředí je nežádoucí, jelikož by při srážení chloridem vápenatým vznikal hydroxid vápenatý a rušil by následnou izolaci. Neutrální šťávu zfiltrujte za sníženého tlaku na Bűchnerově nálevce a přiveďte k varu. Za varu pak šťávu vysrážejte 25% roztokem CaCl2. Vzniklou sraženinu citrátu vápenatého odfiltrujte na skleněné fritě a promyjte horkou vodou (připravte si ji současně při srážení zahřátím v kádince na vařiči). Sraženinu kvantitativně převeďte do čisté kádinky a rozpusťte v 10 mL 30% kyseliny sírové. Citrát
75

vápenatý má daleko větší rozpustnost ve vodě za studena než za horka. Kádinku postavte na vařič a opět zahřejte. Vařte asi 5-10 minut. Kyselina sírová uvolňuje z citrátu vlastní kyselinu citrónovou za vzniku nerozpustného síranu vápenatého. Roztok odstavte, opatrně přidejte asi 20 mL studené destilované vody a nechejte vychladnout. Síran vápenatý odsajte na skleněné fritě. Filtrát přelijte do porcelánové misky a na vodní lázni zahustěte ke krystalizaci. Misku s krystalizačním roztokem nakonec dejte do exsikátoru, odsajte vzduch a nechejte krystalizovat do příštího cvičení.
VYHODNOCENÍ
1) Zapište chemické rovnice všech reakcí, které jste provedli.
2) Vypočtěte výtěžek krystalizace a stanovte tak přibližné množství kyseliny citrónové v jednom citrónu.
ÚKOL č. 19: Rušená krystalizace technické skalice modré
V tomto úkolu použijeme krystalizaci jako čistící metodu. Skalice modrá se v normálním stavu vyskytuje jako pentahydrát, tzn. jedna molekula síranu měďnatého krystalizuje s pěti molekulami vody. Tvoří trojklonné jehlicovité krystaly. Technická skalice bývá často znečištěná síranem železnatým. Přídavkem malého množství kyseliny dusičné do krystalizačního roztoku zoxidujeme dvojmocné železo na trojmocné, to pak během krystalizace zůstává v matečném louhu. Síran železitý má totiž na rozdíl od síranu měďnatého daleko větší rozpustnost. Podobným způsobem můžeme od sebe oddělovat různé soli lišící se svou rozpustností v daném rozpouštědle.
MATERIÁL
Třecí miska s tloučkem, kádinky, vařič, nálevka, kleště, Pasteurova pipeta, stojan s kruhem, filtrační papír, skleněná tyčinka, podložní sklíčko, Bűchnerova nálevka, odsávací baňka, vývěva, miska s drceným ledem, technická skalice modrá, konc. kyselina dusičná.
POSTUP
Na předvážkách odvažte 5 gramů technické skalice modré a jemně utřete ve třecí misce. Pomocí tabulek vypočtěte množství vody potřebné na rozpuštění tohoto množství za laboratorní teploty. Skalici v tomto množství rozpusťte a přidejte několik kapek koncentrované kyseliny dusičné. Roztok na vařiči přiveďte k varu a zfiltrujte. Filtrát zahustěte ke krystalizaci vyvařením. Průběžně provádějte zkoušku: skleněnou tyčinkou kápněte kapku roztoku na podložní sklíčko a sledujte, zda roztok krystalizuje. V momentě, kdy je roztok připraven, vložte kádinku do misky s rozdrceným ledem a vyčkejte, až se vyloučí krystalky CuSO4.5H2O. Produkt odsajte na Bűchnerově nálevce a vysušte mezi kousky filtračního papíru. Matečný louh znovu zahustěte a již popsaným způsobem tak získáte druhý produkt krystalizace.
VYHODNOCENÍ
1) První i druhý produkt po vysušení zvažte a vypočítejte procentové výtěžky krystalizace. Který produkt bude mít větší čistotu?
2) Do kádinky se 100 mL vody jste nasypali po 10 gramech oxidu chromitého, kamence draselno-hlinitého a skalice modré. Pomocí chemických tabulek navrhněte postup, jak by jste tyto tři látky od sebe oddělili pomocí metod popsaných v této úloze.
76

STANOVENÍ BODU TEPLOTY TÁNÍ
Bod tání je teplota, při níž je tuhá látka v rovnováze se svou taveninou. Je to jedna ze základních fyzikálních veličin vedle bodu varu, hustoty, indexu lomu světla, optické aktivity apod. charakterizujících každou látku. Stanovení této veličiny a srovnání s tabelovanou hodnotou je zvláště pro organického chemika nejrychlejší metodou určení čistoty dané látky. Anorganické sloučeniny mají obvykle příliš vysoký bod tání. Čisté látky mají ostrý bod tání. Již nepatrné znečištění látky se projeví značným snížením bodu tání a to i tehdy, jestliže nečistoty mají bod tání vyšší. Kromě snížení bodu tání pozorujeme i zvětšení intervalu bodu tání. Tohoto jevu využíváme ke zkoušení identity dvou látek s týmž bodem tání. Stejná množství obou látek dokonale promísíme a stanovíme pak bod tání (tzv. směsný bod tání). Je-li nezměněn, jsou obě látky identické, je-li ve srovnání s bodem tání složek nižší, jde o různé látky. Mnohé látky se v okamžiku kdy roztají, zároveň rozloží. Rozklad se obvykle projeví ztmavnutím nebo vývojem plynu. Bod rozkladu je zpravidla neostrý a závisí na rychlosti zahřívání.
Prakticky bod taní stanovujeme tak, že malý vzorek látky napěchovaný ve skleněné tenkostěnné kapiláře umístíme do lázně dobře vodící teplo, tj. do kapaliny nebo do kovového bloku, a pomalu zahříváme tak, aby se podmínky co nejvíce blížily rovnovážným. Přitom sledujeme teplotu lázně a pozorujeme látku. Jako teplotu tání označíme teplotu, při níž se objeví kapalná fáze.
Přístroje pro stanovení teploty tání nazýváme bodotávky. Pro látky s teplotou tání nižší než 120°C používáme skleněné bodotávky. Jsou to skleněné nádobky, které bývají plněny kapalinou o vhodné tepelné kapacitě, např. silikonovým olejem, glycerolem apod. Vhodným tvarem nádobky se dosahuje toho, že při pozvolném zahřívání dochází k proudění kapaliny a současnému prohřívání vzorku a teploměru, na kterém je kapilára se vzorkem připevněna. Na obrázku 34 je skleněný P-bodotávek. U látek s vyšší teplotou tání používáme kovové bodotávky, tzv. Thieleho bloky. Jsou to bloky z kovu dobře vedoucího teplo, do kterých jsou vyvrtány svisle tři otvory (obr. 35). Do prostředního otvoru vkládáme teploměr, do zbývajících kapiláry se vzorkem. U kovového bloku je pomalého zahřívání dosaženo tím, že nezahříváme přímo blok, ale tyčku, kterou je blok zároveň upevněn do stojanu. Vzorek pozorujeme v procházejícím světle lampičky upevněné na stojanu za vzorkem.
Obrázek 34: Skleněný P-bodotávek Obrázek 35: Thieleho kovový blok
V laboratorní praxi se často používá speciálních zařízení určených pro práci v mikroměřítku. Jsou to v podstatě upravené mikroskopy s elektricky vyhřívaným stolkem, který je opatřen teploměrem. Na tento tzv. mikrovýhřevný stolek pokládáme vzorek v uspořádání běžném pro mikroskopování a během zahřívání jej pozorujeme v mikroskopu a čekáme na stav, kdy se krystaly změní v rovnovážnou směs pevné a kapalné fáze.
77

ÚKOL č. 20: Stanovte teplotu bodu tání kofeinu
V této úloze ověříte čistotu vámi izolovaného kofeinu z čajového listí pomocí jedné z nejrychlejších a nejspolehlivějších metod. Teplotu bodu tání budete stanovovat pomocí moderního bodotávku firmy STUART SCIENTIFIC. Jeho princip je založen na Thieleho kovovém bloku. Bodotávek je ovládán elektronicky s digitálním výstupem. Lze na něm měřit současně tři vzorky, umístěné v kapilárách. Odečet se dělá okem pomocí zvětšovacího okuláru.
MATERIÁL
Bodotávek STUART SCIENTIFIC, kapiláry, skleněná trubice, kofein čistý, kofein izolovaný ve cvičení č. 4.
POSTUP
Do předem připravených kapilár (úloha č. 7) vložte vzorek vámi izolovaného kofeinu a vzorek čistého kofeinu. Kapiláry plňte následujícím způsobem. Otevřeným koncem kapiláry nahrňte malé množství suchého vzorku, zbytek kolem kapiláry otřete a kapiláru nechejte asi 5x volně padat svisle postavenou trubicí dlouhou asi 1,5 metru na tvrdou podložku. Vzorek látky se tak dobře napěchuje do malého objemu na dně kapiláry. Sloupeček by měl být vysoký asi 2 mm. Zapněte bodotávek hlavním vypínačem. Kapiláry se vzorkem vložte do bodotávku a zkontrolujte, zda je vzorek dobře viditelný. Funkcí MODE nastavte teplotu, na kterou chcete zahřívat (270ºC), a poté tlačítkem START spusťte výhřev. Kolem předpokládaného bodu tání pozorně sledujte oba vzorky a odečtěte hodnoty bodu tání pro čistý i pro izolovaný kofein.
VYHODNOCENÍ
1) Porovnejte obě získané hodnoty bodu tání. V literatuře či na internetu vyhledejte uváděnou hodnotu bodu tání pro kofein a případné rozdíly vysvětlete.
2) Nakreslete vzorec kofeinu a vysvětlete od které základní sloučeniny je odvozen.
Doporučená literatura:
Kašpárek, F. a kol.: Cvičení z laboratorní techniky, Vydavatelství UP Olomouc, 2000. Mareček, A. a Růžička, A.: Laboratorní technika pro nechemické obory, Vydavatelství MU Brno, 1997. Klečková, M. a Šindelář, Z.: Školní pokusy z anorganické a organické chemie, Vydavatelství UP Olomouc, 1993.
78

Laboratorní cvičení č. 10
CHROMATOGRAFIE NA IONTOMĚNIČÍCH
Současnou práci v biochemickém výzkumu a ve studiu přírodních látek si nelze bez chromatografických metod představit. Jejich principem je rozdělování látek mezi stacionární a mobilní fázi na základě rozdílných afinit složek k uvedeným fázím. Mobilní fáze je buď kapalina nebo plyn, stacionární fáze může mít velmi různorodou formu např. částečky tuhé fáze, tenká vrstva kapaliny na pevných částicích, film kapaliny na vnitřní straně kapiláry atd. Objevení principu chromatografie je připisováno ruskému botanikovi M. S. Cvětovi (1872-1919), který rozdělil chloroplastové pigmenty z rostlinných extraktů na sloupci uhličitanu vápenatého.
Chromatografické metody se mohou dělit podle různých kriterií, nejčastější dělení je podle podstaty procesu zodpovědného za separaci, podle skupenství mobilní fáze, způsobu provedení (sloupcová, tenkovrstevná, papírová atd.) nebo podmínek (nízkotlaká, střednětlaká nebo vysokotlaká kapalinová chromatografie). Podle podstaty procesu je můžeme rozdělit do následujících kategorii. Je však třeba mít na paměti, že zejména při dělení biopolymerů (bílkovin, nukleových kyselin) se jedná často o kombinaci dvou nebo více separačních principů. Tak například při rozdělovací chromatografii může docházet také k adsorpci proteinu na povrch sorbentu nebo se uplatňuje síťový efekt porézních sorbentů (gelová permeační chromatografie).
ADSORPČNÍ CHROMATOGRAFIE
Patří k nejstarším a nejrozšířenějším metodám. Pevný adsorbent je obtékán vhodným rozpouštědlem, které unáší analyzovanou směs látek. Na povrchu adsorbentu dochází k různě silné adsorbci těchto látek a tím k jejich rozdělení. Nejčastěji užívanými adsorbenty jsou látky pórovité struktury jako jsou silikagel, Al2O3, CaCO3, hydroxylapatit či aktivní uhlí. Při adsorpci na polárních sorbentech hrají největší úlohu interakce typu ion - dipól a dipól – dipól, zatímco u nepolárních sorbentů to jsou převážně van der Waalsovy síly. Adsorpční chromatografie může být v provedení sloupcovém (CC – column chromatography) nebo tenkovrstevném (TLC - thin layer chromatography).
Zvláštním případem adsorpční chromatografie je hydrofobní chromatografie biopolymerů (zejména proteinů), která využívá jejich schopnosti adsorbovat se na hydrofobní skupiny a povrchy. Vytěsnění adsorbovaných biopolymerů se provádí nejčastěji snížením iontové síly, popř. přídavkem organického rozpouštědla. Jako adsorpční hydrofobní materiály se nejčastěji používají polysacharidové či polyglykolmethakrylátové matrice s navázanými alkylovými řetězci (Sepharosa, Spheron).
ROZDĚLOVACÍ CHROMATOGRAFIE
Principem je dělení látek mezi dvě navzájem nemísitelná nebo omezeně mísitelná rozpouštědla a je charakterizováno rozdělovací konstantou K, která je rozdílná pro jednotlivé složky směsi a liší se pochopitelně i pro různé systémy rozpouštědel. Volbou složení těchto systémů lze ovlivnit účinnost rozdělení složek směsi. Rozdělovací chromatografie je uspořádána tak, že jedna z kapalných fází je zakotvena na inertní pevný nosič (silikagel, filtrační papír, křemelina, škrob) a stává se tak fází stacionární, zatímco druhá fáze - mobilní - přes tuto přetéká. Látky ze separované směsi se rozpouštějí mezi obě fáze. Na pevný nosič se zakotvuje především vodná stacionární fáze, která má k nosiči vyšší afinitu a jako mobilní je pak používaná směs organických rozpouštědel. Častou formou rozdělovací chromatografie je papírová chromatografie (PC – paper chromatography). Vývoj nových sorbentů a automatizace pak daly vzniknout novému a v současnosti jednomu z nejpoužívanějších typů chromatografie – vysokoúčinné kapalinové chromatografie (HPLC – high performence liquid chromatography). Při HPLC se pracuje za vysokého tlaku, nazývá se také často jako vysokotlaká kapalinová
79

chromatografie. Umožňuje několikanásobně rychlejší a citlivější analýzy všech typů látek oproti klasickým sloupcovým metodám.
AFINITNÍ CHROMATOGRAFIE
Je založena na biochemických interakcích, jako jsou interakce enzym – substrát nebo protilátka – antigen, které probíhají s vysokou selektivitou. Princip spočívá v navázání příslušného substrátu či antigenu chemickou reakcí na určitý sorbent, kterým se pak naplní kolona. Enzym nebo protilátka jsou selektivně vychytávány z analyzované směsi a následně mohou být z kolony vymyty. Afinitní chromatografie se ku příkladu využívá k čištění protilátek z krevního séra.
GELOVÁ PERMEAČNÍ CHROMATOGRAFIE (GPC – GEL PERMEATION CHROMATOGRAPHY)
Dělí látky na základě velikosti jejích molekul a blíže se s ní seznámíme ve cvičení č. 11.
CHROMATOGRAFIE NA IONTOMĚNIČÍCH
Chromatografie na měničích iontů neboli ionexová chromatografie je určena pro separaci látek nesoucích náboj. Při výměně iontů se ionty, které jsou elektrostaticky vázány k pevnému a chemicky inertnímu podkladu, reversibilně vyměňují za ionty z roztoku:
R+A− + B− ↔ R+B− + A−
R+A− je měnič iontů s navázaným aniontem A− - anex a B− odpovídá aniontům v roztoku. Měnič kationtů - katex obsahuje záporně nabité skupiny, které reversibilně vážou kationty. Afinita iontů k ionexu není stejná, ale závisí na velikosti náboje a na poloměru hydratovaného iontu. Rozdíly v nábojových vlastnostech, jichž ionexová chromatografie využívá, jsou u biologických látek značné, proto tímto způsobem lze oddělit i látky jinak velmi podobných vlastností. Při vazbě iontu na ionex je však třeba počítat i s dalšími vlivy a to zejména s van der Waalsovými a polárními interakcemi. Síla vazby je také ovlivněna iontovou silou a hodnotou pH prostředí. Dále je třeba si uvědomit, že látky nesoucí stejný náboj jako ionex nebudou zachyceny vůbec.
Jako chromatografický materiál slouží pro ionexovou chromatografii různé inertní matrice, které mají na svém povrchu kovalentně navázané kladně nebo záporně nabité funkční skupiny (viz tab. 9). Charakter funkčních skupin udává sílu ionexu, jejich celkové množství a využitelnost určuje kapacitu ionexu. Označení slabé, střední a silné ionexy vyjadřuje stupeň disociace skupin v závislosti na pH. Zatímco silné ionexy jsou úplně disociovány v širokém rozmezí pH, disociace slabých ionexů je naopak na pH silně závislá. Slabé katexy ztrácejí náboj při pH pod 6 slabé anexy při pH nad 9.
ANEXY FUNKČNÍ SKUPINA
Aminoethyl (AE-)
Diethylaminoethyl (DEAE-)
Kvarterní aminoethyl (QAE-)
OCH2CH2NH3+ Cl-
OCH2CH2N+H(CH2CH3)2 Cl-
OCH2CH2N+(C2H5)2CH(OH)CH3 Cl-
KATEXY
Karboxymethyl (CM-)
Fosfo (P-)
Sulfopropyl (SP-)
OCH2COO− H+
PO4H2− H+
CH2CH2CH2SO3− H+
Tabulka 9: Funkční skupiny ionexů.
80

Jako matrice se v ionexové chromatografii používají tři druhy materiálů: syntetické pryskyřice, jako je např. nejčastěji používaný polystyren zesíťovaný divinylbenzenem, celulosa a konečně gely na bázi polyakrylamidu, dextranů nebo agarosy.
Hlavní fáze ionexové chromatografie jsou ekvilibrace ionexu, navázání látek ze vzorku a vymytí nenavázaných složek, změna podmínek, která vede k selektivní desorpci, a konečně regenerace ionexu (obr. 36).
Ekvilibrace ionexu se provádí startovním pufrem před tím, než je na kolonu aplikován vzorek. Výběru druhu pufru i pH je třeba věnovat značnou pozornost. Zdaleka ne všechny pufry jsou pro ionexovou chromatografii vhodné a to i v případě, že je splněna podmínka vhodného pH. Iontová síla startovního roztoku, který používáme při prvním stupni chromatografie, by neměla být příliš nízká, protože ekvilibrace ionexu je obtížnější a zdlouhavá, zpomaluje se výměnná kinetika, čímž se zhoršuje chromatografické rozlišení. Dochází rovněž ke smršťování ionexu a tím se mění průtok kolonou. Obvykle se doporučuje iontová síla 0,05 – 0,1 M.
1. 2. 3. 4. 5.
+ + + + ++ + + + ++ + + + +
+ + + + ++ + + + ++ + + + +
protiiontystartovacího pufru separované látky gradientové ionty
Obrázek 36: Průběh ionexové chromatografie. 1 - ionex v rovnováze s protiionty (ekvilibrace ionexu), 2 - náhrada protiontů nabitými skupinami ze separované směsi, 3 - ionty s malou afinitou k ionexu jsou desorbovány a nahrazeny protionty z pufru, 4 - desorpce zbývajících iontů změnou podmínek (náhrada gradientovými ionty), 5 - náhrada gradientových iontů protionty startovního pufru (regenerace ionexu).
Vlastní separační proces probíhá ve dvou oddělených fázích. V prvním kroku dochází k sorpci (navázání) látky, určené k separaci na ionex. Ve druhé fázi musí být navázané látky z kolony desorbovány. Ionexová chromatografie se obvykle provádí buď vsádkově nebo v kolonách. Je třeba vzít v úvahu, že rozměry kolony, resp. rozměry chromatografického materiálu v koloně, mají zásadní význam pro úspěšnost separačního procesu. Výšku kolony je třeba zvolit takovou, aby látky ze směsi byly adsorbovány v horní části kolony, nejméně však 1-2 cm od dolního okraje. Velice složité směsi je nutno dělit na delších sloupcích, pro laboratorní měřítko se však většinou vystačí se sloupci do 20 cm. Průměr sloupce závisí na tom, kolik ionexu je nutno do sloupce umístit (při předem zvolené výšce). Průměr sloupce tedy závisí na požadované kapacitě ionexové náplně. Kolony pro ionexovou chromatografii mají obvykle větší průměr a jsou krátké.
Na kolonu aplikujeme vzorek, který by měl být rozpuštěn v ekvilibračním pufru nebo alespoň by měl mít pH odpovídající zvolené hodnotě a nízkou iontovou sílu. Při ionexové chromatografii nezáleží na objemu aplikovaného vzorku, pouze na množství separovaných látek ve vzorku. Jestliže totiž aplikujeme takové množství, že je přesažena kapacita kolony, část separovaných látek se nenaváže a vymyje se z kolony v mrtvém objemu. Ideální je, obsahuje-li vzorek takové množství dělených látek, které odpovídá asi 80-90% kapacity ionexu.
81
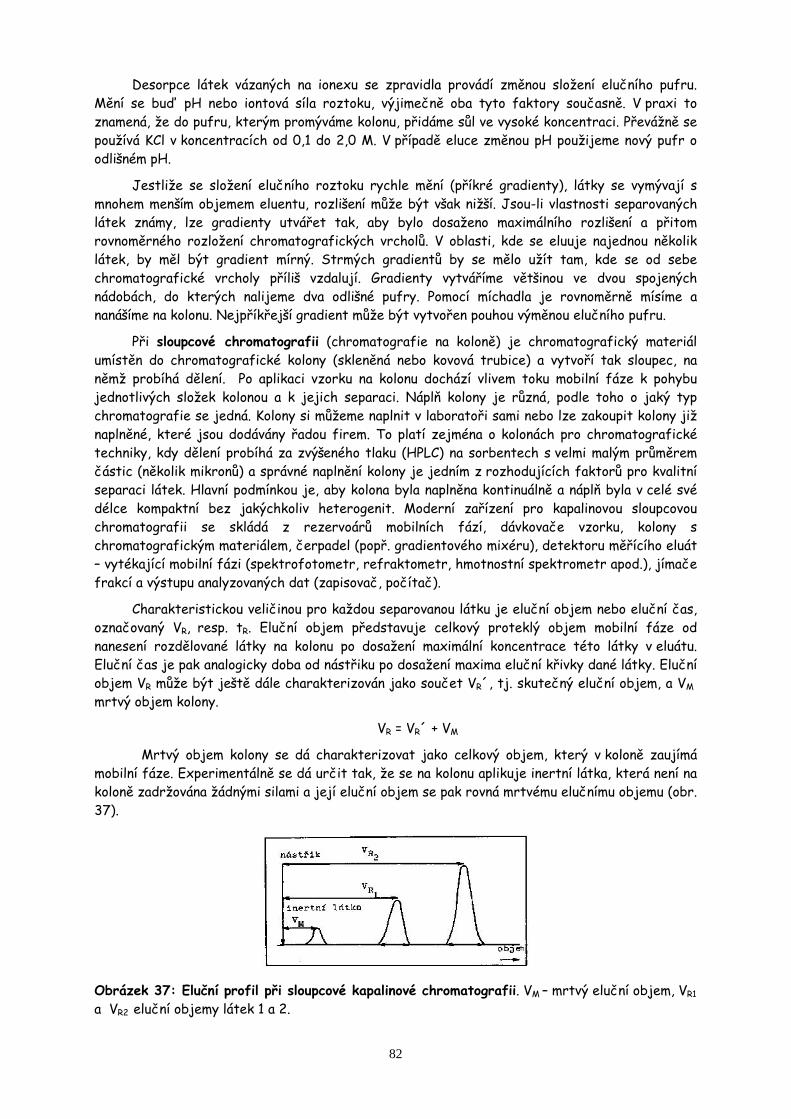
Desorpce látek vázaných na ionexu se zpravidla provádí změnou složení elučního pufru. Mění se buď pH nebo iontová síla roztoku, výjimečně oba tyto faktory současně. V praxi to znamená, že do pufru, kterým promýváme kolonu, přidáme sůl ve vysoké koncentraci. Převážně se používá KCl v koncentracích od 0,1 do 2,0 M. V případě eluce změnou pH použijeme nový pufr o odlišném pH.
Jestliže se složení elučního roztoku rychle mění (příkré gradienty), látky se vymývají s mnohem menším objemem eluentu, rozlišení může být však nižší. Jsou-li vlastnosti separovaných látek známy, lze gradienty utvářet tak, aby bylo dosaženo maximálního rozlišení a přitom rovnoměrného rozložení chromatografických vrcholů. V oblasti, kde se eluuje najednou několik látek, by měl být gradient mírný. Strmých gradientů by se mělo užít tam, kde se od sebe chromatografické vrcholy příliš vzdalují. Gradienty vytváříme většinou ve dvou spojených nádobách, do kterých nalijeme dva odlišné pufry. Pomocí míchadla je rovnoměrně mísíme a nanášíme na kolonu. Nejpříkřejší gradient může být vytvořen pouhou výměnou elučního pufru.
Při sloupcové chromatografii (chromatografie na koloně) je chromatografický materiál umístěn do chromatografické kolony (skleněná nebo kovová trubice) a vytvoří tak sloupec, na němž probíhá dělení. Po aplikaci vzorku na kolonu dochází vlivem toku mobilní fáze k pohybu jednotlivých složek kolonou a k jejich separaci. Náplň kolony je různá, podle toho o jaký typ chromatografie se jedná. Kolony si můžeme naplnit v laboratoři sami nebo lze zakoupit kolony již naplněné, které jsou dodávány řadou firem. To platí zejména o kolonách pro chromatografické techniky, kdy dělení probíhá za zvýšeného tlaku (HPLC) na sorbentech s velmi malým průměrem částic (několik mikronů) a správné naplnění kolony je jedním z rozhodujících faktorů pro kvalitní separaci látek. Hlavní podmínkou je, aby kolona byla naplněna kontinuálně a náplň byla v celé své délce kompaktní bez jakýchkoliv heterogenit. Moderní zařízení pro kapalinovou sloupcovou chromatografii se skládá z rezervoárů mobilních fází, dávkovače vzorku, kolony s chromatografickým materiálem, čerpadel (popř. gradientového mixéru), detektoru měřícího eluát – vytékající mobilní fázi (spektrofotometr, refraktometr, hmotnostní spektrometr apod.), jímače frakcí a výstupu analyzovaných dat (zapisovač, počítač).
Charakteristickou veličinou pro každou separovanou látku je eluční objem nebo eluční čas, označovaný VR, resp. tR. Eluční objem představuje celkový proteklý objem mobilní fáze od nanesení rozdělované látky na kolonu po dosažení maximální koncentrace této látky v eluátu. Eluční čas je pak analogicky doba od nástřiku po dosažení maxima eluční křivky dané látky. Eluční objem VR může být ještě dále charakterizován jako součet VR´, tj. skutečný eluční objem, a VM mrtvý objem kolony.
VR = VR´ + VM
Mrtvý objem kolony se dá charakterizovat jako celkový objem, který v koloně zaujímá mobilní fáze. Experimentálně se dá určit tak, že se na kolonu aplikuje inertní látka, která není na koloně zadržována žádnými silami a její eluční objem se pak rovná mrtvému elučnímu objemu (obr. 37).
Obrázek 37: Eluční profil při sloupcové kapalinové chromatografii. VM – mrtvý eluční objem, VR1 a VR2 eluční objemy látek 1 a 2.
82

ÚKOL č. 21: Rozdělení směsi aminokyselin na silném katexu
Zvláštní, leč v biochemii velmi významnou skupinu látek tvoří ty, jejichž nábojové vlastnosti závisí na pH (aminokyseliny, peptidy a bílkoviny) a tudíž mohou být separovány jak na anexech tak na katexech. Volba ionexu v tomto případě závisí zejména na hodnotě izoelektrického bodu (pI). Izoelektrický bod amfipolární látky je hodnota pH, při které má její molekula nulový náboj. Při pH nižším, než je izoelektrický bod, nese látka kladný náboj a váže se na katex. Při pH nad izoelektrickým bodem má záporný náboj a váže se tedy na anex. U aminokyselin to znamená, že při pH nižším, než je pI, dává molekule náboj skupina NH3
+ a při pH vyšším pak skupina COO-. Při dělení proteinů je třeba brát také v úvahu jejich stabilitu v závislosti na pH. Izoelektrický bod proteinu je součtem izoelektrických bodů všech aminokyselin v něm obsažených.
V této úloze budete dělit směs tří aminokyselin, které se značně liší hodnotou svého pI na sloupci silného katexu. Aminokyseliny se navážou na katex v silně kyselém prostředí, kdy má většina z nich záporný náboj a postupně se budou vymývat vzrůstající hodnotou pH elučního pufru. V eluátu budete aminokyseliny detekovat pomocí ninhydrinového činidla. Barevnou reakci s ninhydrinem poskytují všechny volné aminokyseliny. Aminoskupina aminokyselin poskytuje s ninhydrinem červeno-fialové zabarvení po zahřátí na 100ºC, kromě prolinu a asparaginu, které dávají žluté zabarvení. Arginin budete dokazovat specifickou Sakaguchiho reakcí, kdy jeho guanidinová skupina reaguje s bromnanem a α-naftolem v alkalickém prostředí za vzniku malinově červeného produktu.
MATERIÁL A CHEMIKÁLIE
Skleněná kolona (25 cm) s gumovou zátkou a hadičkou, stojan se svorkou, kádinky, pH metr, rozprašovač aerosolu, filtrační papír, odměrná baňka, Pasteurovy pipety, kulatý stojan na zkumavky, stříkačka, zkumavky, odměrný válec, kapkovací destička, elektrický fén, sušárna, automatická pipeta, skelná vata Akuver, 50 mM citrátové pufry podle McIlvaine pH 3 a 6, 50 mM Tris/HCl pufr pH 9, katex Amberlite IR 120 v citrátovém pufru pH 3, 2,5% vodný roztok serinu, valinu a argininu (směs), 2,5% vodný roztok argininu, 0,4% etanolický roztok ninhydrinu, alkalický roztok bromnanu sodného, etanolický roztok α-naftolu, 10% roztok hydroxidu sodného.
POSTUP
Příprava kolony: Skleněnou kolonu s kohoutem upevněte vertikálně do stojanu. Na dno kolony umístěte chomáč skelné vaty Akuver tak, aby pokryla celé dno. Regenerovaný katex Amberlite IR 120 rozmíchejte asi ve dvojnásobku objemu citrátového pufru, pH 3 a nalijte do kolony, jejíž kohout je otevřený. Sledujte rychlost, s níž se katex usazuje, a neustále dolévejte nové podíly ionexu tak, aby nedošlo k vytvoření vrstev katexu. Katex musí tvořit jeden kompaktní sloupec. Až sahá sloupec asi 3 cm pod ústí kolony, nechejte katex pořádně usadit a pufr odpusťte asi 1/2 cm nad hladinu katexu a kohoutem kolonu uzavřete. Při chromatografii nikdy nenechejte sloupec náplně vyschnout! Poté umístěte na kolonu provrtanou gumovou zátku s hadičkou, která vede do litrové odměrné baňky umístěné nad kolonou obsahující zásobní eluční pufr – citrátový pufr pH 3. Stříkačkou nasajte pufr tak, aby kontinuálně protékal. Zátkou uzavřete kolonu a dole ji kohoutem otevřete. Pod výtok z kolony umístěte 50 mL kádinku a promývejte kolonu citrátovým pufrem, pH 3 tak dlouho, až hodnota pH eluátu dosáhne hodnoty pH 3. Eluát z kolony průběžně proměřujte na pH metru.
Nanesení vzorku: Před nanesením vzorku na kolonu vypusťte veškerý pufr nad sloupcem kolony a kolonu uzavřete. Poté opatrně automatickou pipetou naneste na kolonu 1 mL směsi aminokyselin a kolonu otevřete. Po vsáknutí vzorku do kolony automatickou pipetou naneste na kolonu 3 mL elučního pufru – citrátový pufr, pH 3 a nechejte vsáknout. Poté opět napojte zátku s hadičkou ze zásobního rezervoáru.
Sběr frakcí eluátu: Od okamžiku nanesení vzorku na kolonu měřte pomocí odměrného válce pod kolonou objem eluátu. Do kruhového stojanu si připravte sadu 10 kalibrovaných zkumavek
83

označených 1 až 10 a po odkapání 10 mL eluátu z kolony jímejte frakce po 5 mL do jednotlivých zkumavek.
Detekce eluátu: Aby jste zjistili, ve kterých zkumavkách jsou aminokyseliny, odeberete z každé zkumavky automatickou pipetou 20 µL vzorku a necháte je po kapkách vsáknout do filtračního papíru (3 × 10 cm), kde budete mít čísly označené body jako frakce. Kapku vždy vysušíte elektrickým fénem. Její průměr by neměl přesáhnout 1 cm. Po vysušení papír v digestoři postříkáte aerosolem ninhydrinového činidla z rozprašovače a dáte do sušárny zahřát na 10 minut při 110 oC. Frakce obsahující aminokyseliny se zbarví červeno-fialově. Pokud je i desátá skvrna intenzivně zabarvená ve vymývání a detekci budete pokračovat. V této fázi jsou ze směsi odděleny kyselé aminokyseliny.
Pokud už nejsou v posledních či dalších zkumavkách aminokyseliny, uzavřete kohout a v zásobní odměrné baňce vyměníte pufr za citrátový pufr, pH 6 a necháte ho protéct hadičkou až k zátce. Poté zátkou uzavřete znovu kolonu a do čistých zkumavek budete jimát frakce 11-20 opět po 5 mL. Po najímání všech frakcí proměříte a zapíšete pH ve zkumavkách a provedete detekční test na aminokyseliny s ninhydrinem. Pokud již 20 zkumavka neobsahuje aminokyseliny vyměníte eluční pufr za 50 mM Tris/HCl pufr, pH 9 a jímáte další frakce 21-30, které obsahují bazické aminokyseliny. Opět proměříte pH jednotlivých zkumavek a uděláte test na aminokyseliny.
Během celé chromatografie provádějte na kapkovací destičce test na arginin. Vždy v rozmezí zhruba 20 vytečených mililitrů z kolony odeberete do jamky na kapkovací destičce asi 4 kapky eluátu, přidáte k nim jednu kapku 10% NaOH, 2 kapky etanolického roztoku α-naftolu a kapku alkalického bromnanu. Reakci si nejprve vyzkoušejte s jednou kapkou zásobního roztoku argininu. U pozitivní reakce si poznačte zhruba objem vytečeného eluátu od začátku chromatografie.
VYHODNOCENÍ
1) Sestrojte eluční profil chromatografie (chromatogram) – závislost objemu vytečeného eluátu na pH a vyznačte místa, ve kterých z kolony vytékaly aminokyseliny.
2) Rozhodněte, která z aminokyselin (valin, arginin, serin) vytékala jako první, druhá a třetí a proč? Pomoci vám může specifický test na arginin, popř. literatura.
3) Napište iontové formy všech tří aminokyselin, které existují ve vodných roztocích o pH 2, 6 a 10.
ÚKOL č. 22: Regenerace ionexu
Po skončené eluci se obvykle provádí regenerace ionexu. V malých analytických pokusech je regenerace někdy neekonomická, protože množství ionexu v koloně je velmi malé a práce spojená s regenerací je nákladnější. Pracuje-li se však ve větším měřítku, je třeba regeneraci provést. Jestliže již byly eluovány všechny látky nanesené na kolonu, uskuteční se regenerace promytím startovním pufrem. Kontrolu ekvilibrace provedete tak, že změříme pH a vodivost ve vytékající kapalině. Jestliže však některé látky zůstanou zadrženy v koloně, je třeba je vymýt. Chcete-li tak učinit u látek navázaných silami iontové povahy, zvýšíte iontovou sílu. K odstranění látek, které na koloně vyprecipitovaly nebo jsou velmi pevně vázány, se dá použít 0,1 M NaOH, detergentů, které se na ionex neváží, nebo organických rozpouštědel, jako je např. ethanol.
Většina ionexů, kromě gelových, se však musí po každém použití znovu nastavit do požadovaného cyklu (zregenerovat). Jinak jejich kapacita rapidně klesá. Živcové ionexy se střídavě promývají v 1-4 M roztocích HCl a NaOH, přičemž mezi jednotlivými cykly se vždy nadbytečné ionty vymyjí destilovanou vodou do neutrální reakce. U celulosových ionexů se používají mírnější podmínky - maximálně 0,5 M roztoky kyseliny či zásady. Pořadí kyseliny a
84

zásady je vždy takové, že anex se nejprve promyje v roztoku kyseliny a katex hydroxidu. Po vymytí vodou se pak anex regeneruje v hydroxidu a katex v kyselině, po vymytí nadbytečných iontů se ionexy stabilizují v pufru, ve kterém začíná dělení.
MATERIÁL A CHEMIKÁLIE
Vývěva, skleněná nálevka s fritou, odsávací baňka, kádinky, skleněná tyčinka, pH papírky, pH metr, konc. kyselina chlorovodíková, hydroxid sodný, citrátový pufr pH 3, použitý katex z kolony Amberlite IR 120 (z předchozího cvičení).
POSTUP
Připravte si 100 mL 1 M roztoku NaOH. Použitý katex odsajte na fritě a přeneste do roztoku hydroxidu. Za občasného míchání skleněnou tyčinkou nechejte stát katex v roztoku hydroxidu asi 1/2 hodiny. Roztok hydroxidu poté dekantujte a promývejte několikrát destilovanou vodou. Nakonec odsajte a na fritě ještě několikrát promyjte vodou až do neutrální reakce. Pomocí pH papírku analyzujte výtok ze stopky frity. Připravte si 100 mL 1 M roztoku HCl a katex do něj převeďte a nechejte za občasného míchání stát při laboratorní teplotě. Po 1/2 hodině opět dekantujte a na fritě promývejte až do neutrální reakce. Poté katex ekvilibrujte v citrátovém pufru, pH 3, a to tak dlouho, až je jeho pH totožné s pH pufru.
Poznámka:
Roztoky hydroxidů připravujte vždy tak, že budete přidávat pevný hydroxid do vody. Reakce je silně exotermická, a kdyby jste lili vodu přímo na hydroxid, mohlo by dojít k přehřátí skla a jeho prasknutí.
Stejně tak postupujte při ředění kyseliny, přidávejte ji vždy do vody.
Ionexy a ostatní strukturované náplně chromatografických kolon nikdy nemíchejte na elektromagnetických míchadlech, mohlo by dojít k jejich narušení.
Doporučená literatura:
Káš, J. a kol.: Laboratorní cvičení z biochemie, Nakladatelství Olomouc, 2000. Ferenčík, M. a kol.: Biochemické laboratórne metódy, Alfa Bratislava, 1981. Anzenbacher, P. a Kovář, J.: Metody chemického výzkumu pro biochemiky, VN MON Praha, 1986. Voet, D.A. a Voet, J.G.: Biochemie, Academia Publishing Praha, 1995.
85
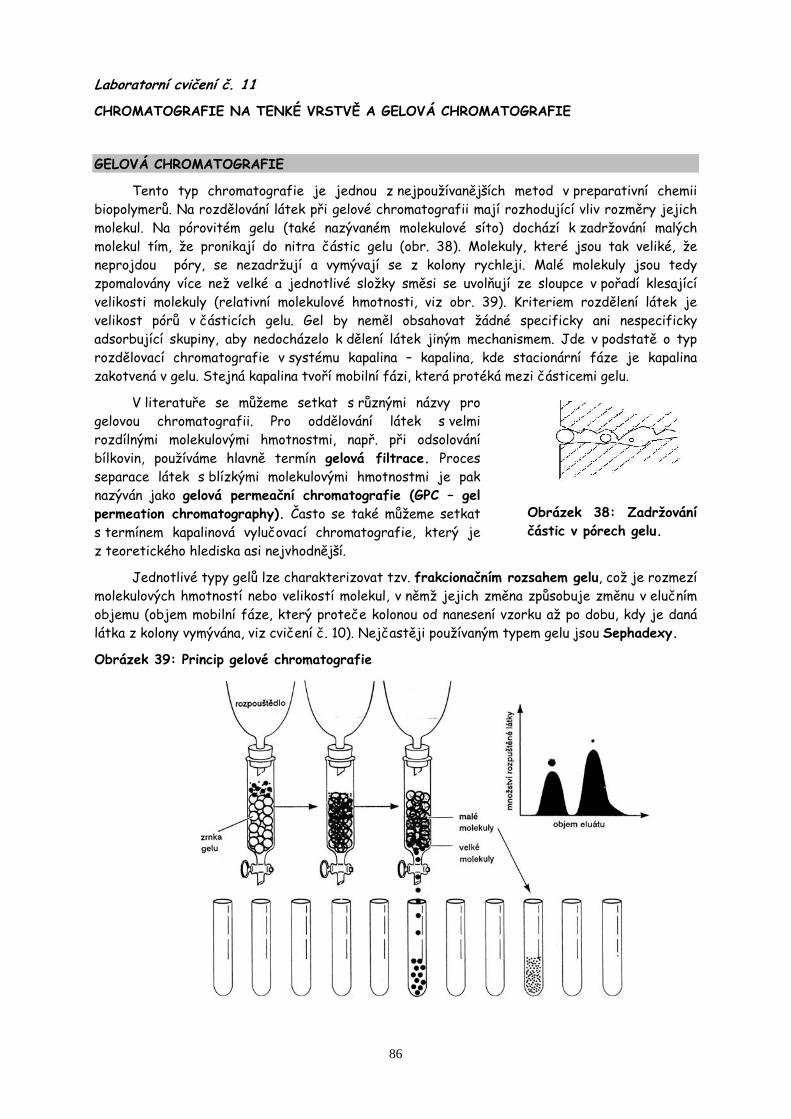
Laboratorní cvičení č. 11
CHROMATOGRAFIE NA TENKÉ VRSTVĚ A GELOVÁ CHROMATOGRAFIE
GELOVÁ CHROMATOGRAFIE
Tento typ chromatografie je jednou z nejpoužívanějších metod v preparativní chemii biopolymerů. Na rozdělování látek při gelové chromatografii mají rozhodující vliv rozměry jejich molekul. Na pórovitém gelu (také nazývaném molekulové síto) dochází k zadržování malých molekul tím, že pronikají do nitra částic gelu (obr. 38). Molekuly, které jsou tak veliké, že neprojdou póry, se nezadržují a vymývají se z kolony rychleji. Malé molekuly jsou tedy zpomalovány více než velké a jednotlivé složky směsi se uvolňují ze sloupce v pořadí klesající velikosti molekuly (relativní molekulové hmotnosti, viz obr. 39). Kriteriem rozdělení látek je velikost pórů v částicích gelu. Gel by neměl obsahovat žádné specificky ani nespecificky adsorbující skupiny, aby nedocházelo k dělení látek jiným mechanismem. Jde v podstatě o typ rozdělovací chromatografie v systému kapalina – kapalina, kde stacionární fáze je kapalina zakotvená v gelu. Stejná kapalina tvoří mobilní fázi, která protéká mezi částicemi gelu.
V literatuře se můžeme setkat s různými názvy pro gelovou chromatografii. Pro oddělování látek s velmi rozdílnými molekulovými hmotnostmi, např. při odsolování bílkovin, používáme hlavně termín gelová filtrace. Proces separace látek s blízkými molekulovými hmotnostmi je pak nazýván jako gelová permeační chromatografie (GPC – gel permeation chromatography). Často se také můžeme setkat s termínem kapalinová vylučovací chromatografie, který je z teoretického hlediska asi nejvhodnější.
Obrázek 38: Zadržování částic v pórech gelu.
Jednotlivé typy gelů lze charakterizovat tzv. frakcionačním rozsahem gelu, což je rozmezí molekulových hmotností nebo velikostí molekul, v němž jejich změna způsobuje změnu v elučním objemu (objem mobilní fáze, který proteče kolonou od nanesení vzorku až po dobu, kdy je daná látka z kolony vymývána, viz cvičení č. 10). Nejčastěji používaným typem gelu jsou Sephadexy.
Obrázek 39: Princip gelové chromatografie
86

Sephadexy jsou vyráběny z fragmentů dextranu, což je polysacharid s relativní molekulovou hmotností okolo 107 – 108, který vzniká ze sacharosy působením bakterie Leuconostoc mesenteroides. Dextranová vlákna se pak zesíťují příčnými vazbami působením epichlorhydrinu (obr. 40). Stupeň zesíťování a s tím související velikost póru lze ovlivnit vzájemným poměrem epichlorhydrinu a dextranu. Jednotlivé typy Sephadexů se označují číslicí a písmenem. Na odsolování se nejčastěji používají Sephadexy s nižším číslem. Pro dělení látek o vyšších molekulových hmotnostech Sephadexy G-75, G-150 nebo jiné typy gelů, jako je Sepharosa, Sephacryl, Bio-gel atd.
Obrázek 40: Část zesíťované struktury Sephadexu
Gelové permeační chromatografie se velmi často užívá pro stanovování relativní molekulové hmotnosti makromolekul. Před vlastní chromatografii daného vzorku je nutno provést separaci standardů o známé relativní molekulové hmotnosti a z jejich elučních objemů sestrojit závislost. Většinou vynášíme závislost retenčního času na logaritmu molekulové hmotnosti. Z retenčního času vzorku o neznámé hmotnosti, který je chromatografován za stejných podmínek jako směs standardů, pak jednoduše odečteme jeho relativní molekulovou hmotnost. Podmínkou je, aby vzorek nebyl znečištěn látkami o přibližně stejné molekulové hmotnosti, které zkreslují retenční čas vzorku. Důležité je také vhodné zvolení typu gelu tak, aby se do jeho frakcionačního rozsahu vešly všechny standardy a samozřejmě i vzorek. Na obrázku 41 je znázorněn eluční profil směsi standardů na koloně Superosy 12 HR používané v systému FPLC (fast protein liquid chromatography).
Odsolování roztoků biopolymerů pomocí gelové chromatografie (gelová filtrace) bývá často vhodnější než dialýza. Odsolováním nestabilních biopolymerů dialýzou dochází někdy k jejich denaturaci z důvodu časové náročnosti procesu. Používá se nejčastěji Sephadex G-25, pro odsolování biopolymerů s relativní molekulovou hmotností nad 30. 000 je pak vhodnější Sephadex G-50. Důležitá je také délka kolony, při větších objemech nanášeného vzorku je nutno úměrně zvyšovat délku náplně v koloně tak, aby se během průtoku směs nízko a vysokomolekulárních látek stačila od sebe oddělit. Obecně se udává, že při gelové chromatografii by měl činit objem nanášeného vzorku maximálně 1-2% z celkového objemu kolony. Pro gelovou chromatografii se proto používají kolony 75 cm dlouhé a často i delší.
87

Obrázek 41: Typický chromatogram směsí proteinů na koloně Superosy 12 HR. 1 – protilátka IgG (Mr 160.000), 2 – hovězí sérový albumin (Mr 67.000), 3 – β-laktoglobulin (Mr 37.000), 4 – cytochrom c (Mr 12.400), 5 - vitamin B12 (Mr 1.355), 6 - cytidin (Mr 246).
CHROMATOGRAFIE NA TENKÉ VRSTVĚ
Tato varianta provedení chromatografického pokusu se nejčastěji provádí u adsorpční, případně u rozdělovací a ionexové chromatografie. TLC používáme především pro analýzu zkoumané směsi, v menší míře pro preparativní účely. Hlavní využití má TLC při separaci nepolárních látek.
Pro tenkovrstvou chromatografii se používají sypané vrstvy (např. oxid hlinitý, celulosa), které jsou však kvůli mechanické stabilitě méně vhodné než vrstvy lité. Adsorbent je nanášen na podložku ve stejnoměrné vrstvičce pomocí skleněné tyčinky s gumovými zarážkami (viz obr. 42).
Obrázek 42: Příprava sypané tenké vrstvy. Obrázek 43: Vyvíjení sypané vrstvy
v horizontální poloze. A – deska s vrstvou, B – rozpouštědlo.
Lité vrstvy se připravují ze suspenze stacionární fáze (např. silikagel) a pojidla (např. sádra). Nejpoužívanější jsou komerčně dostupné lité vrstvy Al2O3 na hliníkové fólii známe pod názvy Silufol či Alufol. Cenově dražší jsou vrstvy, kdy je jako nosič použito sklo a na něj upevněn silikagel nebo celulosa. S oblibou se používají i polymerní nosiče s nanesenou vrstvou polyamidu.
88

Vlastní provedení tenkovrstvé chromatografie může probíhat v několika uspořádáních. Klasické je šikmé uspořádání, kdy desku s tenkou vrstvou umístíme do vyvíjecí nádoby (chromatografická komůrka) asi pod úhlem 20 stupňů a na dno nádoby nalijeme rozpouštědlo (obr. 43). Důležité je, aby chromatografická komůrka byla uzavřena a atmosféra uvnitř se mohla nasytit párami rozpouštědla. Pokud je deska umístěná v komůrce vertikálně, mluvíme o chromatografii vzestupné nebo sestupné, v závislosti na tom, zda je rozpouštědlo umístěno dole nebo nahoře. Vzestupná chromatografie se provádí ve speciálních komůrkách, ve kterých lze vyvíjet i více chromatogramu najednou (obr. 44). Při sestupné chromatografii se jako nosič většinou používá speciální chromatografický papír (komerčně dodávaný pod značkou Whatman), který se zavěsí do nádobky s rozpouštědlem (obr. 45).
Obrázek 44: Vyvíjecí komora pro větší počet desek.
Připomeňme jen, že papírová chromatografie je založená na principu rozdělovací chromatografie, na rozdíl od výše uváděných chromatografii na tenkých vrstvách, které především fungují na principu adsorpční chromatografie. Papírová chromatografie ve vertikálním uspořádání se provádí většinou jako kruhová. Doprostřed papíru umístíme knot, kterým vzlíná rozpouštědlo a kolem něj nanášíme vzorky, které pak migrují k okraji papíru (obr. 46).
Obrázek 45: Zařízení pro sestupnou Obrázek 46: Kruhové vyvíjení papírové papírovou chromatografii chromatografie
Velkou předností chromatografie na tenkých vrstvách je velká časová úspora, malá
spotřeba látek i rozpouštědel, minimální experimentální zařízení a snadné provedení. Jelikož při TLC a papírové chromatografii nemůžeme přímo zjistit charakteristickou veličinu udávající mobilitu dané látky – eluční objem či retenční čas jako u sloupcové chromatografie, používá se pro charakterizaci a identifikaci látek jiná veličina, tzv. retenční faktor Rf. Retenční faktor udává poměr vzdálenosti středu skvrny látky od startu (VM) ku vzdálenosti čela mobilní fáze (VR), tj. vzdálenost kam až doputuje mobilní fáze. Rf = VM / VR

Při separaci směsi látek je vždy žádoucí provádět i chromatografii standardů v tomtéž pokusu. Identifikace je pak snadnější. Kvantitativní vyhodnocení se provádí denzitometricky nebo vyškrabáním a následnou elucí jednotlivých skvrn (změřením koncentrace látek v eluátech).
Detekce látek při tenkovrstevné a papírové chromatografii se provádí různými způsoby:
− přímá – skvrny barevných látek jsou dobře vidět pouhým okem (např. rostlinná barviva),
− absorpce světla v UV oblasti – používá se adsorbentu obsahující fluoreskující složku, po osvětlení UV světlem deska fluoreskuje pouze v místech rozdělených látek jsou vidět tmavá místa,
− fluorescence – některé látky po ozáření UV světlem samy svítí (např. některé alkaloidy),
− specifické barvení – chromatogram se obvykle vyvíjí postříkáním nějakým specifickým činidlem, které se separovanou látkou tvoří barevné produkty, např. ninhydrin je specifické činidlo na aminokyseliny, se kterými dává fialově-červené produkty,
− izotopové techniky – látky jsou označeny radioaktivními izotopy a při vyhodnocování se měří vznikající radioaktivita.
ÚKOL č. 23: Chromatografie karotenoidů z papriky na tenké vrstvě
Karotenoidy jsou rostlinná barviva rozpustná v tucích, v přírodě hojně zastoupená ve všech rostlinných druzích, obvykle zbarvena žlutě, oranžově a červeně. Některé typy karotenoidů se nacházejí i v říši živočišné, dávají například charakteristické zabarvení plameňáků, kanárků, raků nebo třeba slunéčka sedmitečného. Z chemického hlediska je lze řadit do skupiny terpenoidů, tzn. látek odvozených od izoprenové podjednotky. Používají se především jako nezávadná, přirozená barviva v potravinářském a kosmetickém průmyslu. V rostlinném organismu jsou nezbytné pro růst a fotosyntézu, pro člověka jsou nezastupitelným zdrojem vitaminu A, jehož jsou prekurzorem. V současné době se také hovoří o jejich schopnosti snižovat některá rizika rakoviny. Hojně se využívají jako antioxidanty zachycující pro organismus nebezpečné volné radikály.
Lycopen (rajče)
Beta-karoten (mrkev)
Kapsantin (paprika)
90

(kukuřice)
Obrázek 47: Nejznámější typy karotenoidů, jejich vzorce a typicky výskyt.
Extrakt z papriky (Capsicum annum) obsahuje 37 – 54 pigmentů, počet závisí na způsobu
izolace. Většina z nich je na bázi karotenoidů. Hlavním pigmentem papriky je červený kapsantin a kapsorubin, žluto-oranžový beta-karoten a žluté luteiny a xantofyly. Karotenoidy z papriky budete chromatografovat v nepolárním benzenu. Mobilita jednotlivých molekul závisí na jejich polaritě, tzn. především na počtu hydroxy skupin.
POZOR: Pracujete s karcinogenním benzenem. Chraňte se rukavicemi a pracujte v digestoři!
MATERIÁL A CHEMIKÁLIE
Skleněná vana, velká a malá skleněná deska, skleněná tyčinka s gumičkami, odměrné válce, třecí miska s tloučkem, pipety, kádinky, nálevka, filtrační kruh, filtrační papír, vodní lázeň, odpařovací miska, umělohmotné zátky, Silufol, Al2O3 pro chromatografii, benzin, benzen, červená paprika.
POSTUP
Odvážte 1 gram suché papriky a ve třecí misce jej rozetřete s 10 mL směsi benzin -benzen (4:1). Karotenoidy společně s dalšími látkami přejdou do rozpouštědla. Extrakt odfiltrujte přes hladký suchý filtr do kádinky. Pozor, veškeré sklo musí být dokonale suché! Na vodní lázni zfiltrovaný extrakt odpařte do sucha a odparek rozpusťte v 1 mL čistého benzinu.
Příprava sypané tenké vrstvy: Menší skleněnou destičku si položte na stůl a na jeden z jejich konců nasypte hromádku oxidu hlinitého určeného pro chromatografii. Pak opatrně skleněnou tyčinkou na konci s gumičkami projeďte po celé délce skla tak, aby se na jeho povrchu vytvořila jednolitá vrstva oxidu hlinitého. Destičku opatrně chytněte za hrany a přeneste do ploché skleněné vany, na jejíž jeden konec si předem položíte dvě umělohmotné zátky (obr. 43).
Destička je umístěná šikmo pod úhlem asi 20º. Asi centimetr od spodního okraje naneste jako souvislou čáru v délce okolo 3 centimetrů extrakt z papriky. Vzorek nanášejte automatickou pipetou s rozsahem 20-200 mikrolitrů. Na dno nádoby nalijte čistý benzen tak, aby začal vzlínat po destičce nahoru a nádobku překryjte velkým sklem. Chromatogram nechejte vyvíjet a sledujte rozdělování jednotlivých barviv.
Stejný vzorek extraktu z papriky rozdělte vzestupnou chromatografii na vrstvě silufolu. Z folie silufolu si odstřihněte pruh asi o šířce 2 cm. Silufol stříhejte v rukavicích, aby jste ho zbytečně neznečistili. Asi 1 cm od jednoho konce folie nanesete vzorek. Pruh silufolu umístěte do odměrného válce, na jehož dno nalijete mobilní fázi – benzen. Dbejte na to, aby hladina rozpouštědla nezasahovala do naneseného vzorku. Válec uzavřete převrácenou kádinkou a nechejte chromatogram vyvíjet. Jakmile rozpouštědlo vyvzlíná na 1 cm pod horní konec folie, chromatogram vyjměte a nechejte na vzduchu oschnout.
91

VYHODNOCENÍ
1) Porovnejte účinnost obou provedení tenkovrstevné chromatografie a rozhodněte, které dělení je lepší.
2) Vyvinutý chromatogram na silufolu překreslete do protokolu a podle barvy jednotlivých pruhů se pokuste určit, o které karotenoidy jde. V potaz berte i polaritu jejich molekul.
3) Z chromatografie na silufolu vypočtěte retenční faktory pro jednotlivá barviva.
92

ÚKOL č. 24: Gelová chromatografie proteinu s nízkomolekulární látkou
V této úloze budete od sebe oddělovat pomocí gelové filtrace hemoglobin a síran nikelnatý. Průběh gelové filtrace je možno dobře sledovat díky barevnosti obou látek. Nikelnaté ionty jsou zelené. Krevní barvivo hemoglobin má červeno-hnědou barvu. Jde o sloučeninu proteinu – globinu a prostetické skupiny – hemu, na který je vázáno dvojmocné železo. Molekula hemoglobinu je tetramer, to znamená, že se k sobě vážou čtyři základní jednotky globinu s hemem. Celková molekulová hmotnost tohoto tetrameru je 67.000 kDa.
MATERIÁL A CHEMIKÁLIE
75 cm dlouhá kolona na chromatografii, nabobtnalý Sephadex G-25, odměrná baňka 1 L, hadička se zátkou, kádinky, zkumavky, pipety, spektrofotometr Shimadzu UV 1204, konduktometr, kyvety, injekční stříkačka, směs hemoglobinu a síranu nikelnatého.
POSTUP
75 cm kolonu naplněnou Sephadexem G-25 postavte na židli do těsné blízkosti pracovního stolu na jehož horní polici umístíte jednolitrovou odměrnou baňku naplněnou destilovanou vodou. Baňku spojte pomocí gumové hadičky se zátkou s horním ústím kolony a nasajte do ní stříkačkou vodu tak, aby po otevření kolony voda kontinuálně protékala z odměrné baňky přes kolonu. Pod kolonu umístěte kádinku, do které bude voda odkapávat. Kolonu nechejte asi 20 minut promývat vodou.
POZOR:
Nikdy nenechejte kolonu vyschnout!!! Dbejte na to, aby nad sloupcem gelu v koloně bylo vždy alespoň minimální množství vody.
Během chromatografie hlídejte zásobník s vodou, aby v něm bylo vždy dostatek vody, vyhnete se tím nechtěnému vyschnutí kolony.
Při každém vyschnutí sloupce gelu je nutno kolonu vždy znova přeplnit.
Když je kolona dostatečně promytá vodou, zavřete kohoutek kolony, tlačkou zastavte průtok vody v hadičce a oddělejte zátku z kolony. Pak opatrně kohoutkem odpusťte vodu ze sloupce gelu právě tak, aby se klesající hladina vody zastavila na začátku sloupce gelu. Napipetujte opatrně 1 mL vzorku na sloupec gelu a nechejte vsáknout. Pod kolonu umístěte 100 mL odměrný válec. Po vsáknutí vzorku na sloupec naneste 1 mL vody a nechejte vsáknout, totéž ještě jednou s vodou zopakujte a až poté na kolonu opět zapojte hadičku ze zásobníku a nechejte vodu volně protékat. Sledujte dělící se látky na koloně jako dva barevné pruhy. Po odkapání 50 mL vody z kolony začněte jímat do předem připravených zkumavek (25) frakce po 3 mL. Pokud nemáte kalibrované zkumavky, vyznačte si na ně ryskou objem 3 mL. Pokud i po odebrání 25 frakcí stále vytéká z kolony barevný roztok, jímejte do dalších zkumavek až do okamžiku, kdy už eluát není na první pohled zabarven.
Po ukončení chromatografie nechejte kolonu ještě asi 20 minut promývat vodou. Najímané frakce analyzujte. Nejprve proměřte pomocí spektrofotometru Shimadzu absorbance jednotlivých frakcí při 410 nm, což je vlnová délka maximální absorbance hemové skupiny hemoglobinu.
Koncentraci nízkomolekulární látky – nikelnaté soli budete detekovat na základě zvýšené vodivosti roztoku. Pomocí konduktometru proměříte vodivost všech frakcí. Do konduktometrické cely napipetujete vždy přesně 1 mL vzorku a po rysku doplníte destilovanou vodou. Pak do cely ponoříte čidlo konduktometru a na displeji odečtete hodnotu vodivosti v µS.cm-1. Mezi jednotlivými měřeními vždy opláchnete čidlo konduktometru destilovanou vodou.
93

VYHODNOCENÍ
1) Sestrojte chromatogram závislosti elučního objemu na absorbanci při 410 nm a vodivosti. Vyznačte maxima příslušející nejvyššímu výtoku jednotlivých složek. Podařilo se vám od sebe dokonale oddělit nízko- a vysokomolekulární látky?
2) Na koloně Sephadexu G-200 dělíte směs cytochromu c, riboflavinu a pepsinu. Která látka bude z kolony vytékat jako první, která jako druhá a třetí?
Doporučená literatura:
Anzenbacher, P. a Kovář, J.: Metody chemického výzkumu pro biochemiky, VN MON Praha, 1986. Lábler, L. a Schwarz, V.: Chromatografie na tenké vrstvě, Nakladatelství ČAV, 1965. Voet, D. a Voet, J. G.: Biochemie, Academia Publishing Praha, 1995. Káš, J. a kol.: Laboratorní cvičení z biochemie, Nakladatelství Olomouc, 2000. Mikeš, V.: Základní biochemické praktikum, Vydavatelství MU Brno, 1992. Karlson, P.: Základy biochemie, Academia Praha, 1965.
Laboratorní cvičení č. 12
PRÁCE S MIKROSKOPEM
Světelná mikroskopie je základní metodou pozorování ve všech biologických oborech, jejichž předmětem je buňka (tkáň, pletivo, mikroorganismus), protože rozlišovací schopnost této metody odpovídá velikosti buněk a mnohých organel. Světelná mikroskopie využívá k zobrazení světelných paprsků (elektronová mikroskopie proudu elektronů).
Podle osvětlení objektu rozlišujeme:
− mikroskopie v procházejícím světle (světlo prochází pozorovaným objektem).
94

− mikroskopie v dopadajícím světle (světlo dopadá shora na povrch objektu).
Řadíme sem mikroskopii - ve světelném poli
- v temném poli
- fázově kontrastní
- interferenční
- polarizační
- fluorescenční
- v neviditelných paprscích (UV, RTG, IČ)
Podle osvětlení okolí objektu rozlišujeme:
− mikroskopie ve světelném poli (zobrazený objekt má tmavý obrys a nalézá se ve světelném zorném poli). Tato metoda je základní a užívá se nejběžněji.
− mikroskopie v temném poli (světlý objekt je v černém - temném poli). Užívá se pro zvláštní případy.
SLOŽENÍ SVĚTELNÉHO MIKROSKOPU (obr.48)
Mechanické díly: stativ, noha stativu, šroub pro hrubé a jemné zaostření, stolek mikroskopu, vodič preparátu, revolverový měnič objektivů, objímka pro kondenzor, objímka pro filtr. U starých mikroskopů úchytka pro zrcátko, pružinky pro přidržení preparátu.
Optické díly: objektivy, okuláry, kondenzor s aperturní irisovou clonou. U starých mikroskopů zrcátko.
Osvětlovací zařízení: lampa v noze stativu s kolektorovou čočkou. Staré mikroskopy samostatnou mikroskopickou lampu.
Clony: pod kondenzorem aperturní irisová clona, dále polní clona.
Objektiv je základní optický prvek mikroskopu (obr. 49). Vytváří zvětšený převrácený obraz předmětu v horní ohniskové rovině objektu. Tento obraz zvětšuje okulár. Zvětšený obraz je registrován oční sítnicí nebo fotografickým filmem.
ČÍSELNÁ APERTURA
Rozlišovací schopnost objektivu je vyjádřena jeho číselnou (numerickou) aperturou A (apertura = lat. otvor):
A = n . sin ω
kde n = index lomu prostředí mezi objektivem a preparátem (pro vzduch je n = 1, pro imerzní olej a sklo n = 1,5), ω je poloviční otvorový úhel. Je to polovina úhlu ω, který svírají protilehlé krajní paprsky vycházející z předmětového bodu (P) na optické ose mikroskopu a vstupující ještě do objektivu (O). Předmětovým bodem rozumíme kterýkoliv bod či strukturu mikroskopického objektu (obr. 50).
Číselná apertura objektivu má velký praktický význam:
− Určuje rozlišovací schopnosti a hranice užitečného zvětšení.
− Ovlivňuje světelnost mikroskopického obrazu: světelnost obrazu je přímo úměrná čtverci A a nepřímo zvětšení objektivu
95

− Ovlivňuje hloubku ostrosti. Snížením A objektivu se zvětšuje tloušťka zobrazené vrstvy preparátu. Objektivy s velkou hodnotou A mají malou penetrační (pronikací) schopnost, zobrazují velmi tenkou vrstvičku. Pro svůj zásadní význam je údaj A uveden na každém objektivu vedle údaje zvětšení.
Obrázek 48: Binokulární mikroskop.
Složení mikroskopu
okulárytrinokulárníhlavice
nosič „flourescenčníchkostek“
port pro kamerurtuťová výbojka
Halogenová žárovka propozorování vprocházejícím světle
nosič objektivů - revolverobjektivy
stolekkondensor
aperturní clona
polní clona
kolektor osvětlovače
LBD filtrND filtr
Obrázek 49: Objektivy.
96

okulár
objektiv
kondenzoraperturní clona
polní clona
objektParfokalita objektivů
Tloušťkakrycíhoskla
Mechanická tubusovádélka
Zvětšení
Pracovnívzdálenost
Optickákorekce
N.A.
Barevný proužek- zvětšení
preparát
stolek
mechanickátubusovádélka
Obrázek 50: Číselná apertura (A) objektivu závisí na velikosti otvorového úhlu (2 ω) a na indexu lomu (n) media mezi objektem (P) v preparátu (pr) a čelní čočkou objektivu (O). Paprsky vystupující z bodu P pod úhlem větším než 2 ω by se do objektivu nedostaly.
Poznatky pro subjektivní mikroskopii lze souhrnně vyjádřit pojmem rozlišivost mikroskopu (dm). Veličina dm informuje o tom, jaké nejmenší rozměry musí mít předmět, abychom jej při určitém celkovém zvětšení mikroskopu dobře rozlišili svýma očima. Vypočítá se podle vzorce:
327 µm dm = -------------
M kde dm je minimální velikost předmětu v µm, 327 µm je vzdálenost dvou bodů, které průměrně unavené oko rozliší jako dva body ze vzdálenost 250 mm, M je celkové zvětšení mikroskopu. M =
97

M objektivu x M okuláru x zvětšovací koeficient tubusu, který se rovná jedné při dodržení předepsané tubusové délky mikroskopu. Při subjektivním mikroskopování volíme celkové zvětšení tak, aby hodnota rozlišivosti (dm) odpovídala velikosti objektu.
NEJZÁKLADNĚJŠÍ PRAVIDLA MIKROSKOPOVÁNÍ
− Do optické osy mikroskopu nastavíme objektiv o menším zvětšení (4x nebo 10x).
− Osvětlíme zorné pole a upravíme rozestup okulárů.
− Zkontrolujeme čistotu optiky.
− Položíme na stolek preparát a upevníme ho, vyhledáme a zaostříme obraz objektu. Začátečníkům doporučujeme postupovat tak, aby se dívali ze strany na vrchol objektivu a takto, pod kontrolou zraku, jej spouštěli makrometrem až do těsné blízkosti povrchu preparátu. Teprve potom je možno sledovat zorné pole okulárem a současně zvedat tubus až do okamžiku, kdy se objeví nejostřejší obraz.
− Ověříme, zdali vidíme oběma očima (okuláry) stejně dobře.
− Makrometrickým šroubem zachycený obraz doostřujeme mikrometrem.
− Pohybujeme preparátem a pátráme po nejvhodnějším místě pro pozorování. Toto místo posuneme doprostřed zorného pole.
− Pro vlastní mikroskopování vyměníme objektiv podle potřeby za jiný a zkontrolujeme nastavení vhodné apertury. K zaostření pak již stačí mikrometrický šroub.
− Po nastavení potřebného zvětšení upravíme vhodně intenzitu světla a kontrast ovládáním clony, případně kondenzoru.
− Při vlastním studiu preparátů jemně pohybujeme mikrometrickým šroubem a tak zaostřujeme různé roviny v preparátu. Pravou rukou můžeme kreslit.
Formy zobrazení mikroskopických objektů jsou v podstatě čtyři: kresba, mikrofotografie, mikrokinemetografický záznam a videozáznam. Kterákoliv z uvedených forem může být v černobílém nebo barevném provedení.
ÚDRŽBA MIKROSKOPU
Čistění optických částí se provádí vatovými tampónky nebo papírky na optiku stíráním ze středu směrem na periferii nebo ze středu po spirále. Lze použít komerčně prodávané prostředky na čistění optiky nebo směs isopropylalkohol + destilovaná voda (1:1) + kapka 1% roztoku SDS. Čistící směs se nesmí nikdy aplikovat přímo na optiku, ale pouze zvlhčit čistící materiál. Nikdy nepoužívejte korosivní látky a rozpouštědla. Prach se odstraňuje ofukováním balónkem. Z mechanických částí se prach odstraňuje jemným štětečkem.
MIKROSKOPICKÝ PREPARÁT A OBJEKT
Mikroskopický preparát se skládá z podložního skla a krycího skla, mezi skly je medium a v něm je uložen objekt. Preparát bereme zásadně za hrany. Preparáty typu krevního nátěru se mohou pozorovat bez krycího skla. Dobře připravený preparát je předpokladem dobrého zobrazení objektu.
Mikroskopické preparáty dělíme na:
− čerstvé – dočasné (nativní)
− trvalé (pracuje se s materiálem usmrceným, fixovaným, konzervovaným)
98

Čerstvé preparáty umožňují pozorovat pohyb a činnost celých buněk nebo organel. Nevýhodou je omezený výběr materiálu a časové omezení. K přípravě čerstvých preparátů jsou vhodné izolované buňky, části tkání a drobnohlední živočichové. Preparáty prohlížíme ve vodě nebo ve fyziologickém roztoku. Fyziologická media jsou:
− přirozená – krevní sérum, amiová tekutina
− umělá – fyziologický roztok, Ringerův roztok aj.
U čerstvých preparátů používáme často vitální barvení. Rozlišujeme:
− intravitální barvení – barvení živých, zcela neporušených buněk (př. prvoci)
− supravitální barvení – barvení přežívajících buněk vyňatých z těla
− postvitální barvení – barvení odumírajících buněk
Příprava čerstvého preparátu. Do středu podložního sklíčka přeneseme kapátkem kapku vody nebo jiného media, do ní štětečkem, preparační jehlou nebo pinzetou vložíme připravený vzorek. Byla-li kapka malá proniká vzduch pod krycí sklíčko, odstraníme ho přikápnutím vody (media) těsně k okraji krycího sklíčka. Naopak, je-li vody pod krycím sklíčkem více a objekt se pohybuje, musíme přebytečnou vodu odsát filtračním papírem. Dostane-li se voda na svrchní stranu krycího sklíčka je nutné zhotovit nový preparát.
Krycí sklíčko pokládáme vždy nejdříve šikmo na hranu a pak zvolna spouštíme na objekt.
Trvalý preparát je uzavřen do tzv. uzavíracího média, kterým je nejčastěji kanadský balzám. Před uzavřením se musí preparát odvodnit a to řadou alkoholů o vzestupné koncentraci. Trvalé preparáty umožňují nejen lépe rozlišit jednotlivé struktury, ale především slouží jako dokladový nebo demonstrační materiál.
K přípravě tenkých řezů používáme speciálních mikrotomů (obr. 51). Nejjednodušší ruční mikrotom je tvořen dutým válcem na horním konci opatřeným černým hladkým stolkem, na spodním otáčivou hlavicí mikrometrického šroubu, který ve svislém směru pohybuje svorkou umístěnou uvnitř válce. Do ní se pomocí bočního šroubu upevní v bezové duši objekt, který se řeže. Objekty se řežou břitvou. Mokrým štětečkem přenášíme řezy do vody (media).
Obrázek 51: Typy mikrotomů.
Řezán í objektů:– vo lnou rukou
– s pom oc í ručn ího m ikrotom u
– strojn ím m ikrotom e m
– rotačn í
– sáňkové
– konzo lové
99

ÚKOL č. 25: Mikrosublimace kofeinu
Hmota má schopnost přecházet ze skupenství pevného do kapalného či plynného a naopak. Přechod ze stavu pevného do stavu plynného se nazývá sublimací. Této vlastnosti, kterou má mnoho organických látek nacházejících se v léčivých rostlinách, se využívá ve farmakologických laboratořích. Se sublimátem lze provádět řadu mikroreakcí.
Farmakologicky je zajímavý metylovaný xantin kofein - 1,3,7-trimethyl xantin, který je obsažen v kávě i čaji. Kofein je jedním z důležitých léčiv, ovlivňujících krevní oběh.
Kofein lze prokázat tzv. murexidovou reakcí. Jedná se o barevný test na kyselinu močovou a jiné puriny. Pevný vzorek je oxidován v kyselém prostředí, odpařen a následně po přídavku amonných iontů vzniká purpurové zbarvení náležející vznikajícímu murexidu (amonná sůl 5, 5´-nitrildibarbiturová kyselina).
kofein murexid
N
N N
NO
OCH3
CH3CH3
NH
NH
O
O
N
O
NH
NH
O
ONH4
O
MATERIÁL
3x hodinové sklíčko o průměru asi 8-10 cm, filtrační papír, stojan, azbestová síťka, lihový vařič, podložní sklíčka, kapátka, mikroskop, 3 % roztok H2O2, koncentrovanou HCl, čpavek.
POSTUP
Práškový čaj (asi na špičku nože) položte do středu hodinového sklíčka (můžete použít i kofein izolovaný v lab. cvičení č. 4), přikryjte druhým, na jehož vrcholu je asi 3 x 3 cm velký navlhčený kus filtračního papíru určeného ke chlazení. Přes azbestovou síťku zahřívejte sklíčko asi 2 minuty. Horní sklo je možné vyměnit za chladné. Na spodní straně hodinového skla se vytvoří jehličky kofeinu, které budete pozorovat pomocí mikroskopu. Poté proveďte tzv. murexidovou reakci k prokázání přítomnosti kofeinu. Kontrolní reakci proveďte rovněž s firemním preparátem kofeinu. Na hodinové sklo se sublimátem a na sklo s firemním kofeinem kápněte 3 % roztok H2O2
a koncentrovanou HCl. Roztok odpařte a přikápněte roztok čpavku. Purpurové zbarvení vám prokáže přítomnost kofeinu.
VYHODNOCENÍ
Popište tvar a barvu vysublimovaných krystalů kofeinu. Krystaly zakreslete do protokolu. Uveďte zda reakce na přítomnost kofeinu byly pozitivní.
ÚKOL č. 26: Stavba rostlinné buňky z pokožky cibule, plasmolýza, deplasmolýza, plazmoptýza
MATERIÁL
Cibule se zbarvenými suknicemi (v buněčné šťávě obsahují antokyan), žiletka, pinzeta, podložní a krycí sklo, mikroskop, kapátka, 1 M roztok sacharózy, mikroskop.
POSTUP
100

Nařežte si několik čtverečků pokožky (asi 5 x 5 mm) přímo na suknici tak, že ostrou žiletkou provedete nehluboké zářezy. Pinzetou snadno sloupnete vyříznuté čtverečky pokožky, které rychle přenesete na podložní sklo do kapky vody nebo do kapky roztoku sacharózy. Pozorovaný objekt překryjte opatrně krycím sklíčkem. Po ukončení pozorování tkáně v roztoku sacharózy přikápněte na jednu stranu krycího sklíčka kapku vody a k druhé straně přiložte proužek filtračního papíru, který vysaje z prostoru pod krycím sklem roztok sacharózy. Tento úkon zopakujte několikrát do úplného odstranění sacharózy z prostředí okolo pozorované tkáně a jejím nahrazení vodou.
VYHODNOCENÍ
Popište a zakreslete tvar buněk pokožky cibule ve vodném prostředí. Buňky jsou v jednom směru protáhlé, mají velkou centrální vakuolu. Buněčná stěna je stejnoměrná silná a mezibuněčné prostory nejsou vyvinuty, což souvisí s krycí funkcí pokožkových buněk. Jádro se nachází při buněčné stěně a je bochníkovitého tvaru.
Zakreslete a popište vliv roztoku sacharózy na pokožkové buňky. V prostředí sacharózy dochází k plasmolýze, která je způsobena tím, že buňky se nacházejí v hypertonickém prostředí a voda z vakuol je odsávána. Vakuola se zmenšuje a cytoplasma se odchlipuje od buněčné stěny. Po přidání destilované vody dochází k deplasmolýze, buňka se nachází v hypotonickém prostředí. Voda vniká do vakuoly, která se zvětšuje a cytoplasma se dostává do původní polohy. Po déle trvajícím pozorování mohou buňky praskat a červenofialový obsah vytéká do okolní vody. Vlivem velkého osmotického tlaku dochází k prasknutí cytoplazmatické membrány i buněčné stěny.
ÚKOL č. 27: Histochemická reakce na vápník
MATERIÁL
Suchá suknice cibule, kádinka, 96% etanol, mikroskop, podložní a krycí sklo, žiletka, pinzeta, kapátko, 5% roztok šťavelanu amonného v 10% kyselině octové, 3% kyselina sírová.
POSTUP
Na malé kousky nařezanou suknici cibule vložte do 96 % etanolu (připraví vedoucí cvičení). Asi po 12 hodinách vložte preparát do vody a pozorujte pod mikroskopem, zda-li jsou přítomny nějaké krystaly. Poté vodu vysajte filtračním papírkem a Pasterovou pipetou opatrně z druhé strany krycího sklíčka přidávejte roztok kyseliny sírové. Za malou chvíli se vyloučí krystalky síranu vápenatého. Na podložní sklíčko kápněte roztok šťavelanu amonného v kyselině octové a do něj umístěte několik kousků suknice, přikryjte krycím sklem a pozorujte vyloučené krystaly.
VYHODNOCENÍ
Popište a zakreslete krystaly šťavelanu vápenatého a síranu vápenatého. Krystaly šťavelanu vápenatého se rychle v prostředí kyseliny sírové rozpouští a na různých místech preparátu se tvoří krystaly síranu vápenatého. Napište rovnici charakterizující tuto reakci. Jaký typ krystalů jste našli v preparátu ve vodném prostředí?
ÚKOL č. 28: Histochemická reakce na lignin
Lignifikace je proces, při kterém se do celulózní kostry buněčné stěny ukládá lignin. K nejznámnějším histochemickým reakcím, které umožňují identifikovat lignin, patří reakce s floroglucinolem a reakce se síranem anilínu. Tyto reakce nejsou pro lignin specifické, nýbrž jsou reakcemi uvedených činidel s koniferylaldehydem a jinými příbuznými látkami, které jsou stavebními kameny ligninu.
101

MATERIÁL
Stonek semenáčků hrachu, kukuřice či fazolu, mikroskop, podložní a krycí sklo, pinzeta, kapátko, štěteček, ruční mikrotom, žiletky, bezová duše, floroglucinolové činidlo (1g rozpustíme v 50 mL ethanolu), 75% roztok glycerolu v 10% kyselině sírové, 25% HCl.
POSTUP
Uřízněte asi 3 cm špalíček bezové duše a rozřízněte ho napůl. Rukojetí špachtle do jeho středu udělejte oboustranně žlábek. Do žlábku vložte 3 cm část stonku (segment) a bezovou duši se segmentem upněte do ručního mikrotomu. Žiletkou mikrotomu připravte co nejtenčí příčné řezy, které vložíte do vody. Po ukončení přípravy preparátu přeneste štětečkem jednotlivé řezy do několika kapek floroglucinolu v Petriho misce. K řezům v misce přikápněte 1 kapku 25% HCl. Asi po 2-5 minutách přeneste vybarvené řezy do roztoku glycerolu v kyselině sírové na podložním skle, překryjte krycím sklíčkem a pozorujte mikroskopem. Pro kontrolu si nechejte několik řezu jen ve vodě a porovnejte s nabarvenými řezy.
VYHODNOCENÍ
Zdřevnatělé buněčné stěny obsahující lignin se na řezu zbarví červeně až červenofialově. Popište a zakreslete svá pozorování. Pomocí anatomického atlasu nebo jiné literatury pojmenujte buňky, u kterých byla prokázána přítomnost ligninu.
Doporučená literatura:
Nováček, F.: Praktikum z rostlinné cytologie a histologie se základy mikroskopické techniky, Vydavatelství UP Olomouc, 1982. Knoz, J. a Opravilová, V.: Základy mikroskopické techniky, Vydavatelství MU Brno 1992. Ruzin, S. E.: Plant microtechnique and microscopy, Oxford University Press, 1999.
102

Laboratorní cvičení č. 13
PRÁCE S MIKROORGANISMY VE STERILNÍM PROSTŘEDÍ, IZOLACE BAKTERIÁLNÍ DNA A AGAROSOVÁ ELEKTROFORESA DNA
V současné době se moderní biochemický výzkum neobejde bez základních znalostí molekulární biologie a metod používaných pro práci s mikroorganismy a rekombinantní DNA. Pojmem rekombinantní DNA rozumíme takovou molekulu DNA, která se nevyskytuje v přírodě přirozeně, ale vznikla zásahem člověka, který její primární strukturu (pořadí deoxyribonukleotidů) upravil tak, aby měla vlastnosti, kterých se dá využít ve výzkumu či při průmyslových výrobách. Rekombinantní DNA se nejčastěji využívá v genovém inženýrství, kde např. vhodným zásahem do promotorových sekvencí genů můžeme mnohonásobně zvýšit expresi genu v umělém expresním systému (baktérie, kvasinka) a získat tak velké množství rekombinantního proteinu, které nelze získat standardními postupy izolace z rostlinného pletiva či živočišné tkáně. V současné době je takto vyráběna řada proteinů (umělé hormony, doplňky výživy atd.). V biochemickém výzkumu si lze molekulárně-biologickými postupy usnadnit práci např. rekombinantní expresí málo abundantního proteinu (enzymu), který je možné následně použít k imunizaci a přípravě protilátek proti tomuto proteinu. Nebo pomocí cílených mutací primární aminokyselinové struktury proteinů můžeme následně sledovat a určovat aminokyselinová residua, která mají esenciální vliv na funkci proteinu.
Elektroforesa, neboli migrace iontů v elektrickém poli, se velmi často používá pro analytické dělení biologických látek, hlavně pak proteinů a nukleových kyselin. Elektroforesa většinou probíhá na vhodném nosiči jako je papír, celulosa nebo polymerní gel.
Papírová elektroforesa
Při papírové elektroforese se vzorek ve formě tečky nanese na pruh filtračního papíru, jehož konce se namočí do vhodných pufrů (elektrolyt). Po zapojení stejnosměrného proudu putují ionty vzorku určitou rychlostí (která je závislá na charakteru iontu např. na jeho velikosti) k opačně nabitým elektrodám. Po určité době působení proudu vznikají zóny odpovídající jednotlivým složkám vzorku (bandy – proužky), které se vhodnou analytickou reakcí detekují.
Gelová elektroforesa
Gelová elektroforesa patří mezi nejpraktičtější a nejvýkonnější metody používané pro dělení makrokolekul. U běžně používaných gelů, jako je agarosa nebo polyakrylamid, je možné ovlivnit velikost jejích pórů a tím dosáhnout nejen dělení na základě pohyblivosti v elektrickém poli, ale i na základě velikosti molekuly. Při gelové elektroforese se velké molekuly oproti malým zpožďují, protože na rozdíl od gelové filtrace (chromatografie) neexistuje při elektroforese kolem gelu prostor pro volné rozpouštědlo. Gel je umístěn přímo v elektrickém přístroji a pohyb velkých molekul je proti pohybu malých molekul zpomalen tím, že obtížněji pronikají gelem.
Obrázek 52: Komůrka pro vertikální elektroforesu v polyakrylamidovém gelu.
103

Při polyakrylamidové gelové elektroforese (PAGE) je gel vytvořen polymerací akrylamidu a N,N´-methylenbisakrylamidu. Gel bývá často připraven ve formě tenké obdélníkové vrstvy, ve které je možno současně analyzovat několik různých vzorků (viz obr. 52). V případě dělení proteinů bývá pH gelu kolem 9, aby molekuly měly záporný náboj a pohybovaly se k anodě umístěné ve spodní nádržce. Každý vzorek se rozpustí v minimálním množství relativně hustého roztoku (nanášecí pufr obsahující glycerol), aby se zabránilo smíchání vzorku s pufrem v horní nádržce a nanese se na povrch gelu do předem vytvořené jamky. Při napětí asi 300 V prochází gelem stejnosměrný proud po dobu nutnou k rozdělení makromolekul do řady jednotlivých proužků. Gel se poté z přístroje vyjme a jednotlivé zóny se vhodně detekují. Nejčastějším typem PAGE je elektroforesa v prostředí detergentu dodecylsulfátu sodného tzv. SDS-PAGE. SDS denaturuje proteiny tím, že se na ně pevně váže a mění jejich terciální strukturu do válcovité podoby. Většina proteinů váže SDS ve stejném poměru a získává silný záporný náboj, tzn., že proteiny pokryté SDS mají shodné poměry počtu nábojů na jednotku hmoty a podobný tvar. Následkem toho se proteiny při elektroforese v prostředí SDS dělí pouze na základě své molekulové hmotnosti. Vhodně zvolenou koncentrací akrylamidu v gelu lze pak dosáhnout optimální dělení proteinů o určité velikosti s odchylkou do 5%. Nejčastěji používané koncentrace akrylamidu jsou mezi 8-12%.
Druhým nejčastějším typem gelové elektroforesy je agarosová elektroforesa, která se používá hlavně na dělení látek s velkou molekulovou hmotností (DNA, RNA). Aby byla zajištěná dostatečná pohyblivost takto velkých molekul, musela by se použít velmi nízká koncentrace akrylamidu (pod 3%) a gel by byl velice měkký a manipulace s ním komplikovaná. Agarosové gely jsou i v nízkých koncentracích velmi pevné a koncentrace agarosy v nich je závislá na velikosti dělených nukleových kyselin, od 3% pro fragmenty DNA a RNA mající desítky až stovky párů bází po 0.8% pro fragmenty velikostí tisíců až desetitisíců párů nukleotidových bází. Chemicky je agarosa polysacharid skládající se ze D-galaktosy a 3,6-anhydro-L-galaktosy, který se izoluje z červených řas – ruduch. Nejčastější uspořádaní agarosové elektroforesy je horizontální (obr. 53 a 55), přičemž princip dělení je na rozdíl od proteinů vždy závislý pouze na velikosti nukleové kyseliny, která je sama o sobě negativně nabitá a pohybuje se proto směrem od katody k anodě. DNA však není v agarosovém ani polyakrylamidovém gelu možno vidět. Je proto nutné ji nějakým způsobem obarvit nebo označit. Nejpoužívanějším barvivem pro DNA jsou planární aromatické kationty, jako je např. ethidium nebo akridinová oranž, které se interkalují (vmezeří) do struktury dvoušroubovice a po ozáření UV světlem následně fluoreskují. Proteiny se nejčastěji po elektroforetickém dělení detekují na gelu pomocí barviva Coomassie brilliant blue nebo citlivější metodou založené na redukci stříbra.
Obrázek 53: Schéma nanášení vzorků a princip agarosové elektroforesy: 1. ztuhlý agarosový gel, 2. nanesení standardu molekulových hmotností, 3. nanesení vzorků, 4. zapojení stejnosměrného proudu, 5. průběh elektroforesy, 6. ukončení elektroforesy.
104

Kapilární elektroforesa
Třebaže jsou různé formy elektroforesy v gelech snadné a lehce dostupné, obvykle probíhají několik hodin a je nesnadné je automatizovat a také kvantitativně vyhodnocovat výsledky. Tyto nedostatky lze obejít použitím kapilární elektroforesy, při níž se vzorky makromolekul dělí ve velmi tenkých kapilárách (průměr 1 až 10 µm) zhotovených z křemene, skla nebo plastu. Tyto tenké kapiláry rychle odvádějí teplo, takže mohou být použita elektrická pole s vysokým napětím (obvykle 100 – 300 V na cm), což je asi stokrát více než v případě standardních horizontálních nebo vertikálních elektrofores čas analýzyse tak sníží na několik minut. Rychlé rozdělení také zamezuje difusnímu rozostření proužků. Kapiláry bývají plněny pouze pufrem nebo velice málo koncentrovaným SDS-polyakrylamidovým gelem. Metody kapilární elektroforesy mají velmi vysoký stupeň rozlišení a často bývají zcela automatizovány pro rychlé analýzy velkého počtu vzorků. V současnosti má kapilární elektroforesa největší využití při sekvencování nukleových kyselin (čtení pořadí jednotlivých bází v řetězci DNA), kdy je třeba mezi sebou rozlišovat fragmenty DNA lišící se ve velikosti pouze jedním nukleotidem.
Práce ve sterilním prostředí
Při práci s DNA a mikroorganismy se velice často setkáváme s požadavkem celkově sterilního prostředí. Znamená to, že veškerý materiál a chemikálie musí být zbaveny jakýchkoli kontaminujících mikroorganismů a ostatních nečistot. Mikroorganismy (baktérie a kvasinky) jsou nejčastěji používány k produkci rekombinantních nukleových kyselin či proteinů, kdy je množíme za specifických podmínek v tzv. živných médiích. Živná média jsou roztoky základních živin –aminokyselin, sacharidů, vitamínů atd., které umožňují rychlé množení a růst daného mikroorganismu. Pokud by se dostaly do takto připravovaných médií náhodné mikroorganismy z ovzduší, vody, či naších rukou, docházelo by také k jejich namnožení a znehodnocení experimentálního vzorku. Všechny roztoky s kterými pracujeme, je proto třeba autoklávovat, tzn. sterilizovat vodní parou za zvýšeného tlaku (120°C, 100 kPa). Autoklávovat lze i veškerý materiál zhotovený z chemického skla či porcelánu a plastové pomůcky zhotovené ze speciálního odolného plastu. Autoklávování je nejúčinnější, pokud na roztok či materiál působí pouze horká vodní pára bez vzduchu. Autoklávy jsou proto složité tlakové nádoby, ze kterých je před vlastním zahřátím vody odsán vzduch a následné natlakování je způsobeno pouze vodní parou. Odolný materiál lze také sterilizovat v horkovzdušných pecích či sušárnách (1-6 hodin při 180-200°C).
Roztoky chemikálií (oligosacharidy, antibiotika atd.), u kterých hrozí za zvýšené teploty rozklad, se sterilizují speciálními membránovými filtry. V současné době se nejvíce používají nitrocelulosové filtry s různou velikostí pórů (20 – 100 µm) a průměru. Nitrocelulosové filtry jsou většinou jednorázové a roztoky se jimi filtrují buď použitím negativního (vodní či mechanická vývěva) nebo pozitivního (injekční stříkačka) tlaku. Viry procházejí většinou bakteriálních filtrů.
Rychlé sterilizace lze dosáhnout také UV zářením. Optimální baktericidní účinek má záření o vlnové délce 254 nm. Malé předměty se takto sterilizují po dobu několika minut ve speciálních sterilizátorech které jsou osazeny několika UV zářiči. Celé laboratoře, větší prostory a vzduch v nich se sterilizuje germicidními lampami, které jsou vhodně rozmístěny v prostoru. Vzhledem k tomu, že účinnost UV záření lineárně klesá se čtvercem vzdálenosti od zdroje záření, pro sterilizaci větších prostorů se používá delší doby (vzhledem k tomu, že UV záření je pro živé organismy mutagenní, zapínají se germicidní lampy v laboratořích většinou přes noc, nebo po skončení práce). Nástroje, které se používají během experimentální práce opakovaně (pinzety, očkovací kličky), se sterilizují otevřeným plamenem za předchozího opláchnutí v 70% ethanolu.
105

Při běžném provozu v laboratoři se však postupnému kontaminování prostředí nikdy nevyhneme. Tato kontaminace pak může negativně ovlivňovat některé experimenty citlivé na znečistění běžně se vyskytujícími bakteriemi či plísněmi. Často k takovéto kontaminaci dochází při očkování kultur pokusnými bakteriemi či kvasinkami, otevírání zásobních láhví s živnými médií, roztírání kultur na pokusné plotny či pěstování rostlin a živočišných buněk v in vitro podmínkách. Tyto operace proto většinou provádíme ve speciálních laminárních boxech (též flow boxy). Jsou to ze všech stran ohraničené pracovní plochy, ve kterých je řízené proudění vzduchu, který prochází přes speciální mechanické filtry. Tím se vzduch přítomný v okolí experimentu neustále zbavuje mikroorganismů a pevných nečistot. Laminární boxy mohou být s vertikálním či horizontálním prouděním vzduchu a k jejích běžnému vybavení patří vlastní germicidní lampa, která se zapíná vždy po skončení práce.
Dá se říci, že efektivita práce v molekulárně-biologické laboratoři velmi závisí na dodržování základních návyků pro práci ve sterilních podmínkách. Je tedy nutné využívat všech dostupných prostředků jak zabránit nechtěné kontaminaci. Udržovat čistotu a pořádek, pro úklid využívat jednorázové papírové ubrousky, používat desinfekci na ruce nebo jednorázové vyšetřovací rukavice, při sebemenším podezření na kontaminaci, roztok či materiál zlikvidovat či znova sterilizovat, většinu manipulace s mikroorganismy směřovat do laminárních boxů, v rámci možností používat jednorázový materiál, používat germicidních zářičů po skončení práce.
Izolace nukleových kyselin
Pro izolaci nukleových kyselin z živých organismů byla vyvinuta řada metod, které se od sebe mohou dosti odlišovat. Pro stručnost tohoto textu budou popsány dvě základní. Prvním krokem u všech metod je vždy lýze buněk, nebo pevné tkáně. U buněk většinou stačí rozrušit buněčnou stěnu a membrány, nejčastěji se používají detergenty jako je dodecylsulfát sodný (SDS) nebo Triton X-100. Pro izolaci DNA nebo RNA z rostlinných pletiv nebo živočišných tkání je třeba nejdříve mechanického narušení, to provádíme buď mrazem – tekutým dusíkem nebo různými typy homogenizátorů. Buněčný obsah včetně DNA se z lyzovaných buněk uvolní do extrakčního pufru, který vždy musí obsahovat chelatační činidlo etylendiaminotetraoctovou kyselinu (EDTA), která vychytá veškeré vápenaté ionty z extraktu. Vápenaté ionty fungují jako kofaktory nukleas, enzymů které štěpí nukleové kyseliny, a pokud by tyto enzymy byly během extrakce aktivní, naštěpily by nám veškeré izolované nukleové kyseliny. Do extrakčního pufru se někdy přidávají navíc ještě proteinázy, enzymy štěpící proteiny. Většina DNA je totiž obalená histony a jejich odstranění proteinasou zvýší čistotu izolované DNA.
Jeden typ metod je založen na extrakci směsi fenolu a chloroformu. Fenol se rozpouští ve vodě i v chloroformu, preferuje ale chloroform. Chloroform se jako organické rozpouštědlo s vodou nemísí. Přidáme-li tedy směs fenolu a chloroformu k extraktu ze živé tkáně, veškeré tuky přecházejí do chloroformu, bílkoviny a část polysacharidů se působením organických rozpouštědel vysráží a nukleové kyseliny zůstanou ve vodné fázi. Po intenzivní extrakci se směs zcentrifuguje a vzniklé fáze od sebe oddělí. Vysrážené proteiny a sacharidy vytvoří na rozhraní bílou prstencovitou sraženinu. Pro dokonalé odstranění polysacharidů se někdy používá činidlo cetyltrimethylamonium bromid (CTAB). Pro vysrážení nukleových kyselin z vodné fáze nakonec použijeme dvou a půl násobek objemu absolutního etanolu nebo isopropanol s přídavkem soli. Po intenzivní centrifugaci nám pak nukleová kyselina vytvoří ve zkumavce opaleskující pelet, který ještě několikrát promyjeme etanolem a nakonec rozpustíme ve vodě nebo vhodném pufru.
Novější metody využívají adsorpce nukleových kyselin na sklo v přítomnosti chaotropních solí, jako je třeba guanidin thiokyanát. Extrakt nukleových kyselin se smíchá se silikátovými kuličkami v přítomnosti chaotropní soli. Nukleové kyseliny se naváží na kuličky a ty se pak
106

promývají, tím se zbavujeme nečistot. Nakonec se nukleové kyseliny z kuliček uvolní snížením iontové síly roztoku. Tato metoda je velice rychlá a efektivní a využívá se ve většině komerčních sestavách např. v diagnostických laboratořích v nemocnicích.
V bakteriálních buňkách bývá část genetické informace kromě vlastní chromosomální DNA uložená také v plasmidech. Plasmidy jsou krátké kruhovité úseky DNA nesoucí genetickou informaci, která není esenciální pro život baktérie, např. geny pro rezistenci k různým typům antibiotik. Uměle vytvořené plasmidy slouží v molekulární biologii pro různé klonovací a expresní techniky. Plasmidy z bakteriální kultury nejčastěji izolujeme tzv. alkalickou metodou. Tato metoda je založena na faktu, že většina chromosomální DNA, bakteriálních proteinů a SDS precipituje při neutralizaci silně alkalického roztoku pomocí octanu draselného, kdežto plasmidová DNA zůstává v roztoku. Precipitát odstraníme centrifugací a plasmidovou DNA ze supernatantu vysrážíme etanolem. Pokud je požadována plasmidová DNA velmi vysoké čistoty, lze pokračovat po odstranění chromozomální DNA a proteinů preparační centrifugací v gradientu chloridu cesného. Další možností je precipitace polyetylenglykolem. Izolovanou DNA lze pak skladovat po dobu několika týdnů v lehce zásaditém pufru o malé iontové síle v lednici, nebo dlouhodobě zamraženou na -70°C.
Koncentraci izolované DNA nejčastěji měříme pomocí spektrofotometru. Nukleové kyseliny absorbují nejintenzivněji světlo o vlnové délce 260 nm, takže hodnota absorbance je přímo úměrná koncentraci DNA. Míru kontaminace proteiny pak můžeme určit z poměru absorbancí při 260 nm a 280 nm, kde mají absorpční maximum proteiny. Pokud se tento poměr pohybuje kolem hodnoty 1.8, je izolovaná DNA relativně čistá. Koncentraci DNA pak lze zjednodušeně vypočítat dle vztahu cDNA = A260 /0.02 za podmínky, že jsme předem ze vzorku odstranili specificky RNA, která má stejné absorpční maximum. Daleko citlivější stanovení koncentrace DNA je pomocí fluorimetrie, kdy měříme fluorescenci DNA po smísení se specifickým interkalačním barvivem.
ÚKOL č.29: Příprava agarových ploten
Mikroorganismy můžeme v laboratoři pěstovat buď na pevném kultivačním médiu nebo v kapalném médiu. Oba typy médií musí být sterilní a jejich složení musí vyhovovat požadavkům mikroorganismu na výživu svým pH a osmotickými poměry, musí obsahovat vhodný zdroj dusíku a uhlíku a případně další látky esenciální pro růst mikroorganismu. Hlavní složky kultivačních médií jsou získávány po kyselé nebo enzymatické hydrolýze kaseinu, fermentativní hydrolýze masa (masový výtažek), autolýzou nebo hydrolýzou pekařského droždí (extrakt z kvasinek), působením žaludečních šťáv na rostlinné a živočišné bílkoviny (pepton). Do pevných kultivačních médií přidáváme 1-3% agar, který po autoklavování a ochlazení ztuhne.
MATERIÁL A CHEMIKÁLIE:
Erlenmayerova baňka z připraveným ztuhlým sterilním LB agarem (složení na 100 mL: 1g bakteriálního tryptonu, 0.5 g kvasničného autolysátu, 0.5 g NaCl, 1.5 g agaru, pH 7.0), sterilní Petriho misky, flow box, kahan, mikrovlnná trouba, ampicilin 100 mg/mL sterilizováno přes filtr.
POSTUP:
V mikrovlnné troubě rozpustíme ztuhlý agar v Erlenmayerově baňce a necháme ochladit zhruba na 50-60 ºC. V zapnutém flow boxu si nachystáme čtyři sterilní Petriho misky. Do dvou misek sterilně nalijeme zhruba po 25 mL LB agaru. Do zbylých 50 mL agaru napipetujeme
107

ampicilin ze zásobního roztoku tak, aby jeho finální koncentrace byla 100 mg na litr. Zamícháme krouživým pohybem ruky a rozlijeme do zbylých dvou misek. Při manipulaci s Erlenmayerovou baňkou vždy opálíme její ústí nad kahanem. Po rozlití agaru do všech misek je necháme stát v zadní části flow boxu s mírně pootevřeným víkem, aby odvětrala kondenzovaná voda. Víka každé Petriho misky řádně popíšeme.
ÚKOL č. 30: Kultivace mikroorganismů na agarových plotnách
Prvním krokem každé kultivace (pomnožování) mikroorganismů je jejich přenesení z uchovávacího média nebo přímo z odebraného vzorku do čerstvého živného prostředí, tomuto přenesení říkáme očkování. Provádí se různými metodami, vždy je nezbytné zachovat všechna pravidla sterilní práce tak, aby se do čerstvého média dostal pouze sledovaný mikroorganismus, nikoliv kontaminace ze vzduchu, rukou, dýchacích cest nebo pracovní plochy. Mikroorganismus vždy přenášíme pomocí bakteriologické kličky. Sterilizace kličky se provádí žíháním v plameni kahanu. Celý drátek se vloží do plamene a nechá rozžhavit. Potom se ve svislé poloze nechá vychladnout. Po naočkování je nutno vždy před odložením kličku sterilizovat žíháním. Kličku vsuneme do zkumavky a do očka nabereme kulturu. Kličku vytáhneme, ožíháme hrdlo zkumavky a zátku (kterou stále držíme malíkem pravé ruky) a zkumavku uzavřeme. Mikroorganismus na plotnu přenášíme tak, že víčko se jen mírně nadzvedne a očkem sterilní kličky se lehce přejede po povrchu agaru. Při neznámém množství mikroorganismu v tekuté kultuře vytváříme na plotně tzv. hádka. Plotnu si pomyslně rozdělíme na čtvrtiny a kličkou začneme v jedné čtvrtině od horního okraje do středu kreslit zúžujícího se hádka (viz obr. 54).
Obrázek 54: Roztírání mikro-organismů na agarovou plotnu, tzv. hádek.
Druhého hádka vytvoříme ve vedlejší čtvrtině, tak že při jeho kreslení zajedeme kličkou do struktury hádka předchozího a tím přeneseme část mikroorganismů znovu na kličku. Stejně tak pokračujeme se zbylými dvěmi čtvrtinami. Principem je vyředit mikroorganismy tak, aby se v posledních hádcích od sebe oddělily na jednotlivé buňky, které dají vznik jednotlivým koloniím. Pokud máme mikroorganismy v kultuře značně naředěné, nebo selektujeme málo abundantní mikroorganismy s rezistencí, naneseme celou tekutou kulturu nebo její alikvot do středu plotny umístěné na kolotoči a předem vyžíhanou hokejkou po roztočení kolotoče ji opatrně rozetřeme po celém povrchu plotny.
MATERIÁL A CHEMIKÁLIE:
LB agarové plotny s ampicilinem a bez ampicilinu (složení na 100 mL: 1 g bakteriálního tryptonu, 0.5 g kvasničného autolysátu, 0.5 g NaCl, 1.5 g agaru, pH 7.0), flow box, kahan, sterilní očkovací kličky, hokejka, kolotoč, kultura baktérií Escherichia coli nesoucí plasmid s rezistencí proti ampicilinu a bez rezistence.
POSTUP:
Na jednu agarovou plotnu s ampicilinem rozetřeme metodou hádka kulturu E. coli nesoucí plasmid s rezistencí k antibiotiku, na druhou plotnu s antibiotikem rozetřeme pomocí hokejky 50 µL kultury E. coli bez rezistence k antibiotiku. Na jednu misku bez antibiotika rozetřete metodou hádka kulturu nesoucí plasmid. A na poslední misku bez antibiotika plivněte a sliny rozetřete
108

hokejkou, nebo plotnu pouze otevřete a intenzivně na ní dýchejte a poté zase zavřete. Po nanesení všech kultur na plotny je dejte kultivovat přes noc do inkubátoru na 37ºC. Plotny dávejte do inkubátoru obráceně (víkem dolů), to proto, aby voda která se z agaru neustále vypařuje a kondenzuje na víku, neskapávala zpátky na kulturu mikroorganismu na plotně rostoucí.
ÚKOL č. 31: Izolace plasmidové DNA z E.coli
MATERIÁL A CHEMIKÁLIE:
Kultura E. coli obsahující plasmid s Ampr genem pěstovaná přes noc, Roztok P1 (50 mM Tris/HCl pH 8.0, 10 mM EDTA, autoklávováno a přidáno 0.1 mg/ml Rnasy A), Roztok P2 (0.2 M NaOH, 1% SDS, autoklávováno), Roztok P3 (3 M octan draselný pH 5.5, autoklávováno), Isopropanol a 70 % ethanol vychlazený v mrazničce (-20oC), TE pufr (10 mM Tris-HCl pH 8,0, 1 mM EDTA), mikrozkumavky 1,5 ml (eppendorfky), miska s ledem, stolní centrifuga s rotorem na mikrozkumavky, automatické pipety a špičky.
POSTUP:
1) 1,5 ml podíl kultury E. coli nesoucí plasmid inkubované přes noc přeneseme do mikrozkumavek, centrifugujeme při 5.000 g 1 min (separace buněk od média) a médium odlijeme.
2) Přidáme 0,3 ml roztoku P1, bakteriální pelet suspendujeme pomocí vortexu.
3) Přidáme 0,3 ml roztoku P2, promícháme několikerým převrácením zkumavky, ponecháme stát 5 min při laboratorní teplotě.
4) Přidáme 0,3 ml roztoku P3, promícháme několikerým převrácením zkumavky, ponecháme stát 5 min na ledě.
5) Centrifugujeme při 14.000 g, 10 min, supernatant přelijeme do nových ependorfek.
6) Přidáme 0,5 ml isopropanolu (laboratorní teplota), centrifugujeme při 14.000 g, 10 min, supernatant odlijeme.
7) Sraženinu DNA promyjeme 0,5 ml 70% ethanolu (-20oC), centrifugujeme při 14.000 g, 5 min, supernatant odlijeme, jeho zbytky odstředíme na dno mikrozkumavky a odpipetujeme. DNA ponecháme sušit 10 min ve flowboxu.
8) Sraženinu DNA resuspedujeme pipetováním v 0,04 ml TE.
ÚKOL č. 32: Elektroforesa DNA
MATERIÁL A CHEMIKÁLIE:
1 % agarosa v 1x TAE pufru (0,04 M Tris-kyselina octová, 0,001 M EDTA), zásobní elektrodový pufr 10x TAE (0,4 M Tris-kyselina octová, 0,01 M EDTA, pH 8,0), 50 % glycerol, 50 mM EDTA pH 8, 0,125 % bromfenolová modř, 0,125 % xylenová violeť, 10% Ethidium bromid, λ DNA marker, mikrozkumavky 1,5 ml (eppendorfky), horizontální elektroforetická komůrka, zdroj napětí, AlphaDigiDoc, systém pro digitální fotodokumentaci gelů, mikropipety, špičky, pikofuga
109

POSTUP:
Pod dohledem vedoucího cvičení nebo laborantky si sestavíme v digestoři elektroforetickou komůrku pro agarosový gel v horizontálním uspořádání. Z termostatu na 60 oC vyjmeme rozpuštěnou 1% agarosu v 1x TAE a nalijeme asi 1 cm silnou vrstvu do elektroforetické komůrky. Agarosa se nalévá do prostoru ohraničeného postranními plastovými deskami na nosnou skleněnou desku (viz obr. 55). Ihned přidáme 40 µl 10% ethidium bromidu (POZOR KARCINOGEN, DBÁT BEZPEČNOSTNÍCH PŘEDPISŮ) a promícháme špičkou. Zhruba 1 cm od startu (katoda, černý kabel) zasadíme do agarosy hřeben tak aby zasahoval asi 0,8 cm do hloubky gelu. Gel ponecháme ztuhnout asi 30 min.
Obrázek 55: Nalévání agarosového gelu
Po 30 minutách odstraníme postranní plastové desky a do komůrky nalijeme asi 200 ml elektrodového pufru 1x TAE tak, aby hladina splývala s agarosovým gelem a nakonec pod hladinou odstraníme hřeben. Během tuhnutí gelu si připravíme vzorky: Do tří mikrozkumavek napipetujeme 20, 10 a 5 µl izolované DNA a přidáme pětinu objemu denaturačního barviva, promícháme a v pikofuze necháme sednout na dno mikrozkumavky.
V digestoři opatrně pipetou naneseme jednotlivé vzorky do jamek na startu elektroforesy. Mezi nanášením jednotlivých vzorků, špičku pipety vyplachujeme v elektrodovém pufru přímo v komůrce. Pečlivě si zaznamenáme pořadí a polohu jednotlivých vzorků. Do jedné jamky jako standard molekulových hmotností DNA naneseme 5 µl λ DNA naštěpenou restrikčním enzymem StyI nebo současně restrikčními enzymy HindIII a EcoRI nanesených vzorků (viz obr. 56).
Elektroforetickou komůrku uzavřeme bezpečnostním víkem, připojíme ke zdroji napětí 120 V po dobu asi 20-30 minut. Průběh elektroforézy je možné sledovat podle pohybu pásů barviv ze vzorků. Vypneme přívod proudu, odkryjeme víko a vyjmeme agarosový gel na nosném skle a pořídíme snímek digitální kamerou podle pokynů vedoucího cvičení.
Po celou dobu práce používáme ochranné nitrilové rukavice (fialové) a dbáme předpisů pro práci s ethidium bromidem, který je obsažen v gelu.
VYHODNOCENÍ:
1) Následující cvičení si vezměte své agarové plotny, které byly přes noc inkubovány na 37 oC a popište a zhodnoťte, co na které vyrostlo či nevyrostlo a proč?
2) Podařilo se vám izolovat plasmidovou DNA? Jak by jste zjistili její množství a čistotu?
3) Zhodnoťte výsledek elektroforesy, co jste viděli na gelu po obarvení ethidium bromidem? Pokuste se vysvětlit proč jste viděli při elektroforese plasmidové DNA více než jeden proužek.
110

Obrázek 56: λ DNA (48.5 kbp) naštípaná restrikčním enzymem StyI (vlevo) nebo HindIII a EcoRI (vpravo).
1% agarosa v TAE
standard vlevo: 11 fragmentů - 19.329, 7.743, 6.223, 4.254, 3.472, 2.690, 1.882, 1.489, 925, 421, 74 bp standard vpravo: 13 fragmentů - 21.226, 5.148, 4.973, 4.268, 3.530, 2.027, 1.904, 1.584, 1.375, 947, 831, 564, 125 bp Doporučená literatura: Ráclavský, V.: Úvod do základních metod molekulární genetiky, Vydavatelství UP Olomouc, 1998. Ruml, T.: Laboratoře z genového inženýrství, Vydavatelství VŠCHT, 1997 Voet, D.A. a Voet, J.G.: Biochemie, Academia Publishing Praha, 1995 Jandová, B. a Kotoučková, L. : Praktikum z Mikrobiologie, Vydavatelství MU, 1996.

















