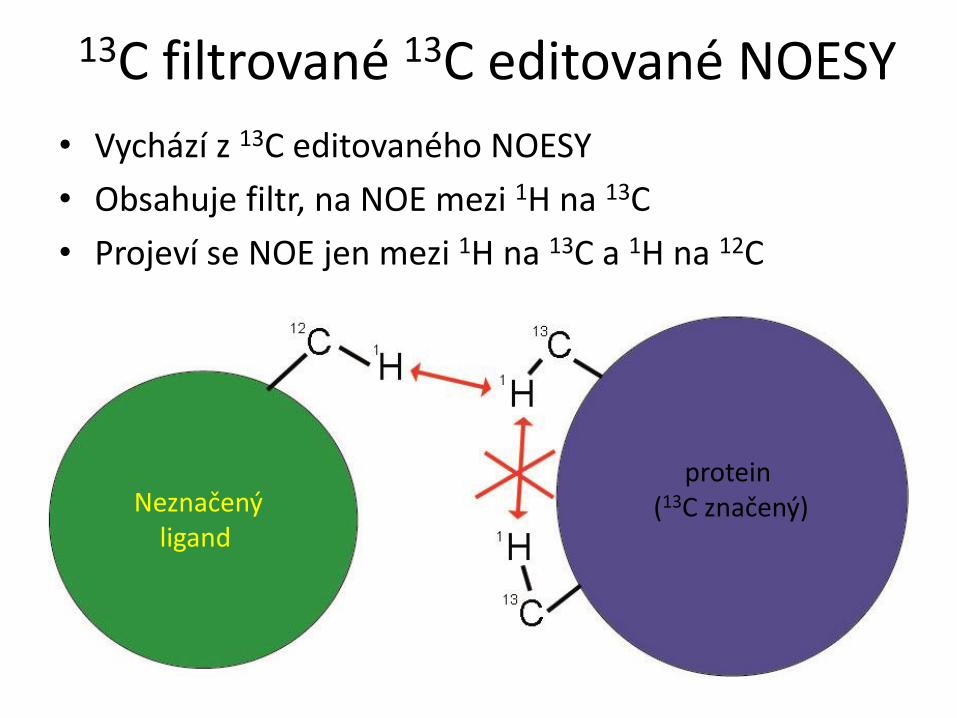
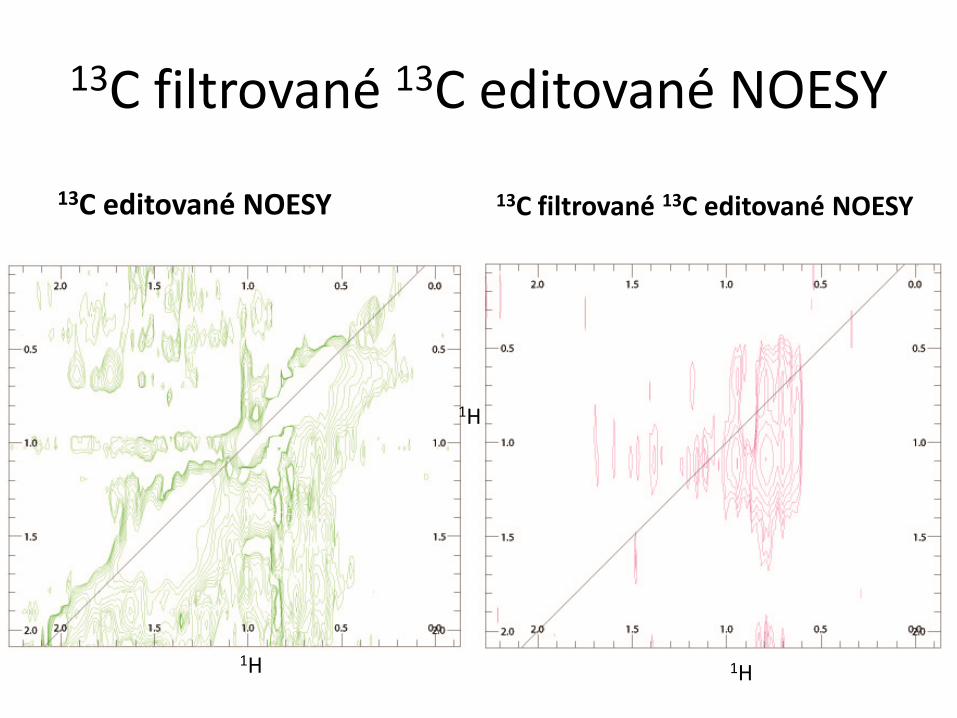
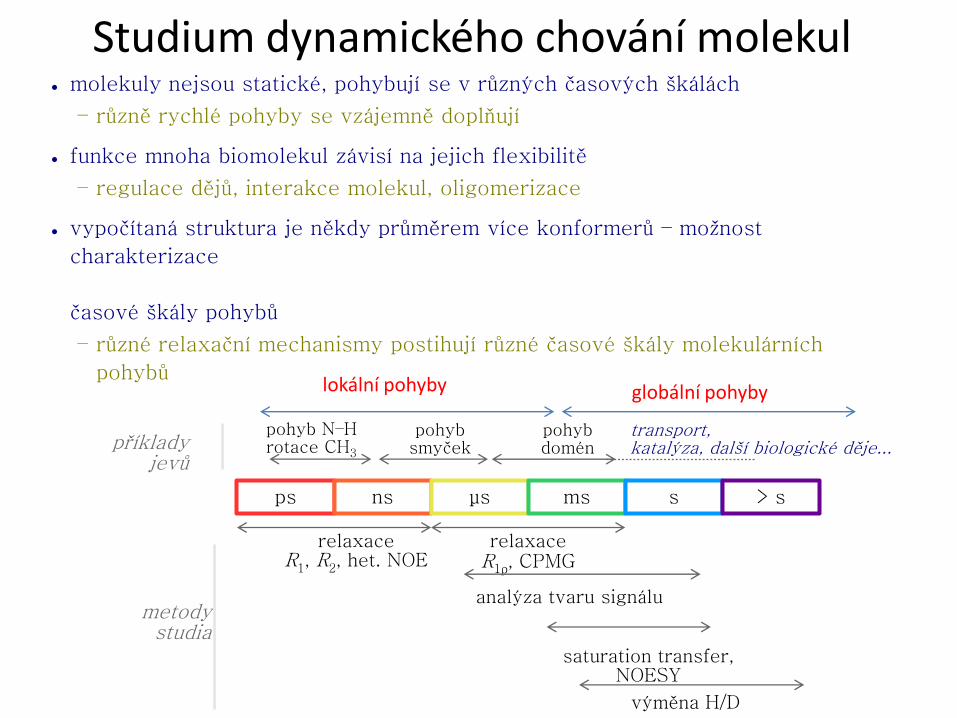
RCSB PDB Progr. NMR RCSB PDB NMR biomakromolekul Typy biomakromolekul a možnosti studia pomocí NMR ● proteiny a peptidy – rozmanité složení, omezení jen velikostí molekul ● nukleové kyseliny (RNA, DNA) a oligonukleotidy – omezení malou rozmanitostí chemického složení ● polysacharidy a oligosacharidy – omezení malou rozmanitostí chemického složení ● kombinace výše uvedených
Transcript
RCSB
PDB
Progr.
NMR
RCSB PDB
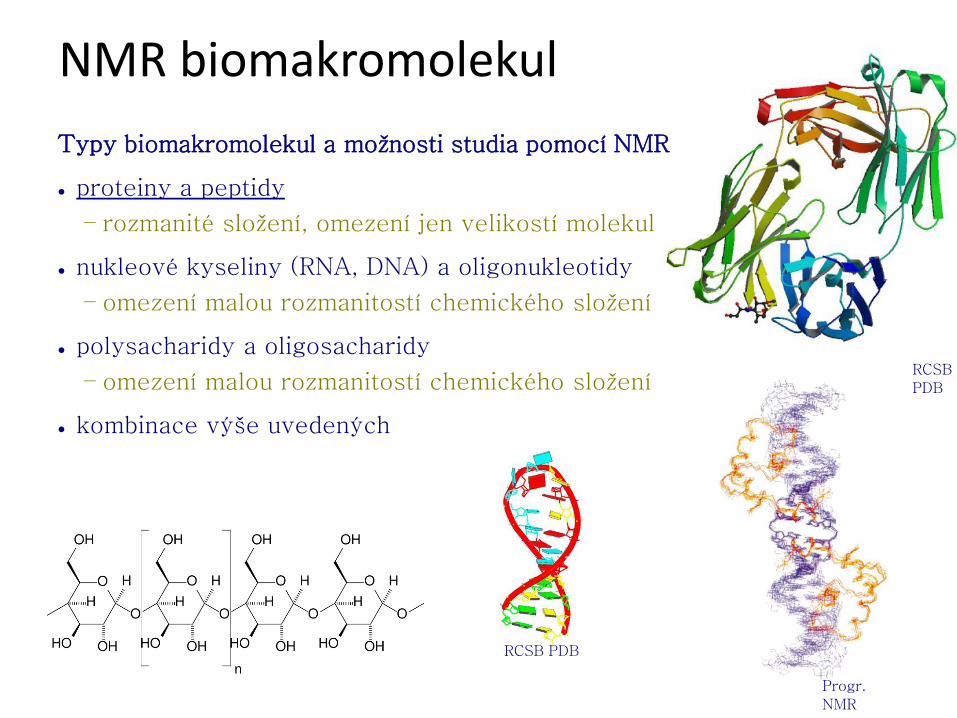
NMR biomakromolekul
Typy biomakromolekul a možnosti studia pomocí NMR
● proteiny a peptidy
– rozmanité složení, omezení jen velikostí molekul
● nukleové kyseliny (RNA, DNA) a oligonukleotidy
– omezení malou rozmanitostí chemického složení
● polysacharidy a oligosacharidy
– omezení malou rozmanitostí chemického složení
● kombinace výše uvedených
NMR Proteinů
• α
•
•
•
•
•
•
• ř
•
•
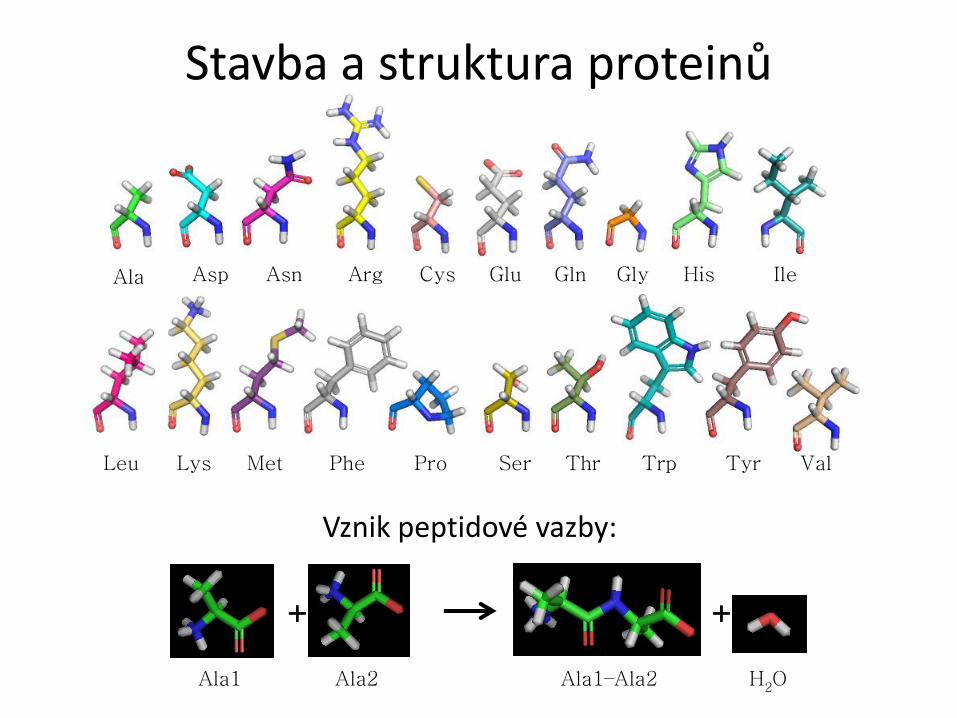
Ala Asp Asn Arg Cys Glu Gln Gly His Ile
Leu Lys Met Phe Pro Ser Thr Trp Tyr Val
Stavba a struktura proteinů
+
Ala1 Ala2 Ala1–Ala2 H2O
+
Vznik peptidové vazby:
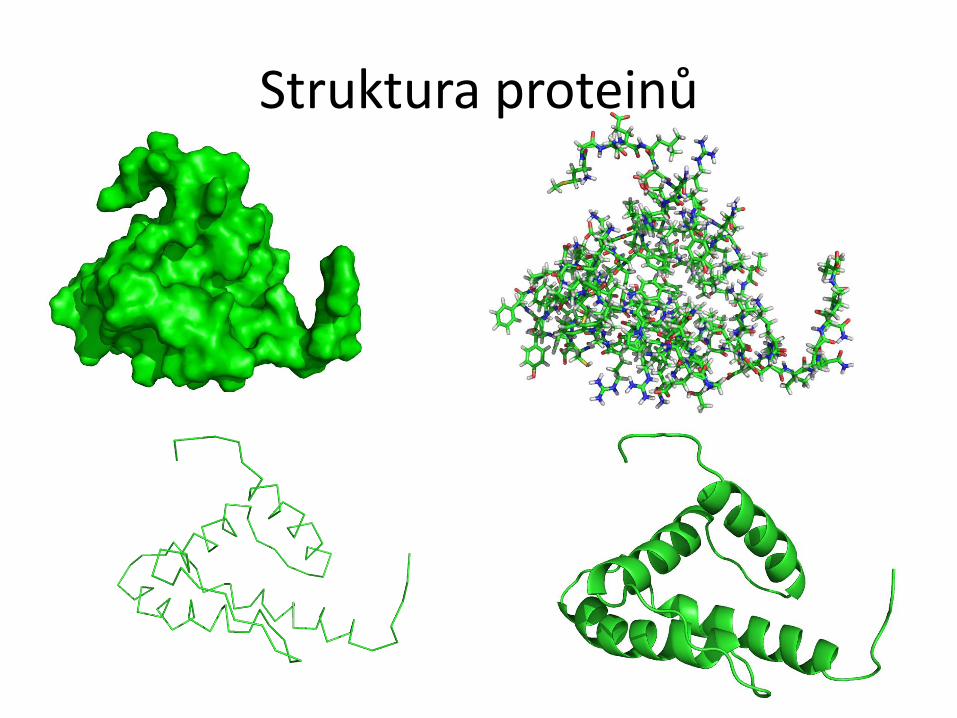
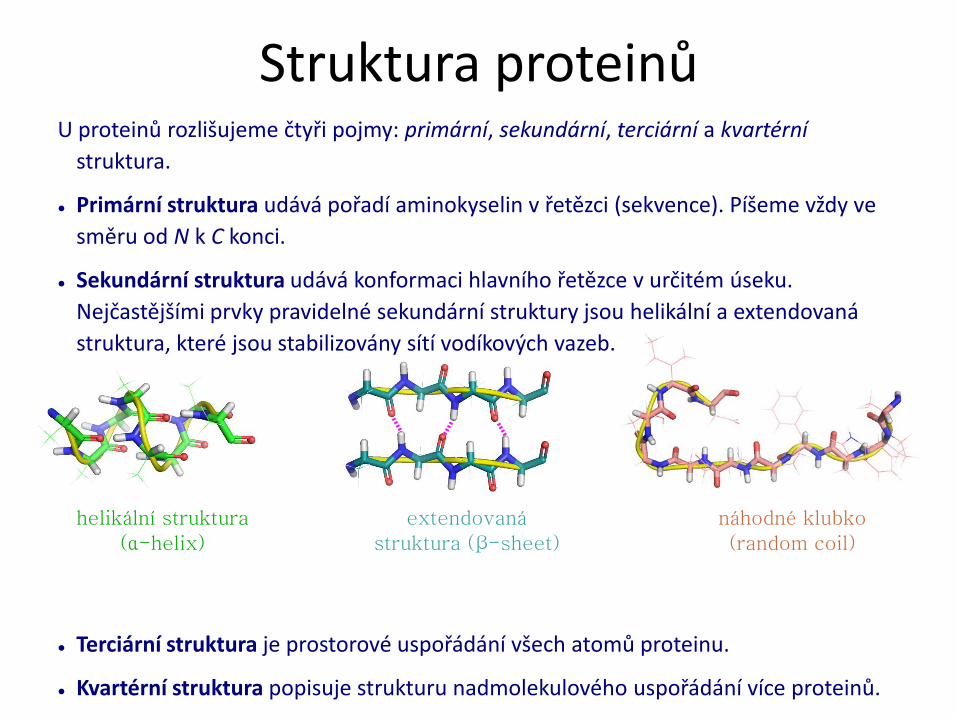
Struktura proteinů
helikální struktura
(α-helix)
extendovaná
struktura (β-sheet)
náhodné klubko
(random coil)
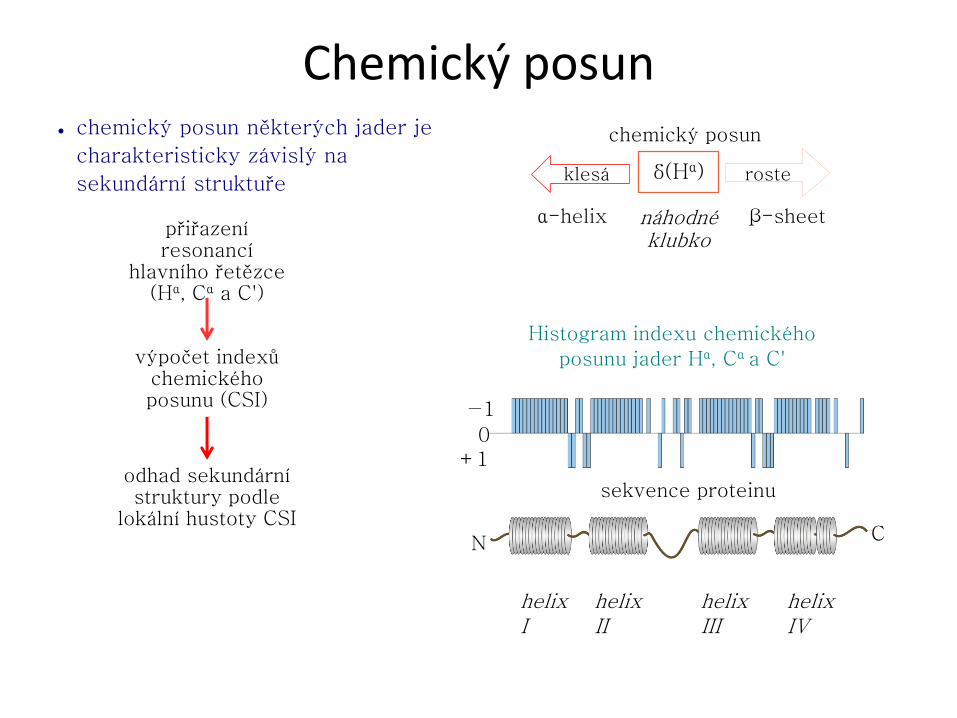
Struktura proteinů U proteinů rozlišujeme čtyři pojmy: primární, sekundární, terciární a kvartérní
struktura.
● Primární struktura udává pořadí aminokyselin v řetězci (sekvence). Píšeme vždy ve
směru od N k C konci.
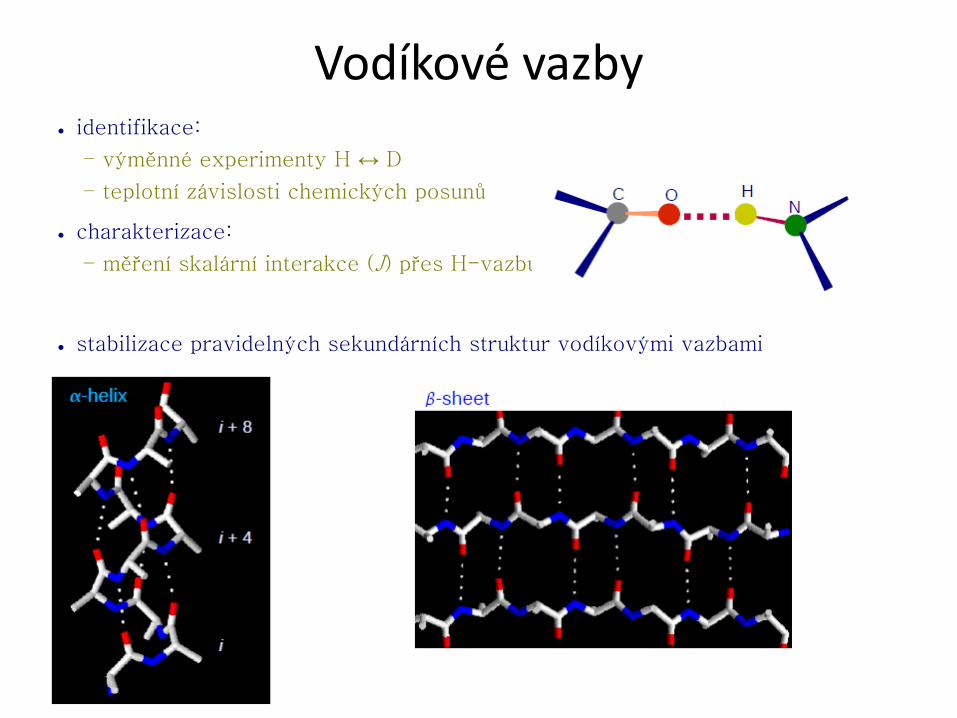
● Sekundární struktura udává konformaci hlavního řetězce v určitém úseku.
Nejčastějšími prvky pravidelné sekundární struktury jsou helikální a extendovaná
struktura, které jsou stabilizovány sítí vodíkových vazeb.
● Terciární struktura je prostorové uspořádání všech atomů proteinu.
● Kvartérní struktura popisuje strukturu nadmolekulového uspořádání více proteinů.
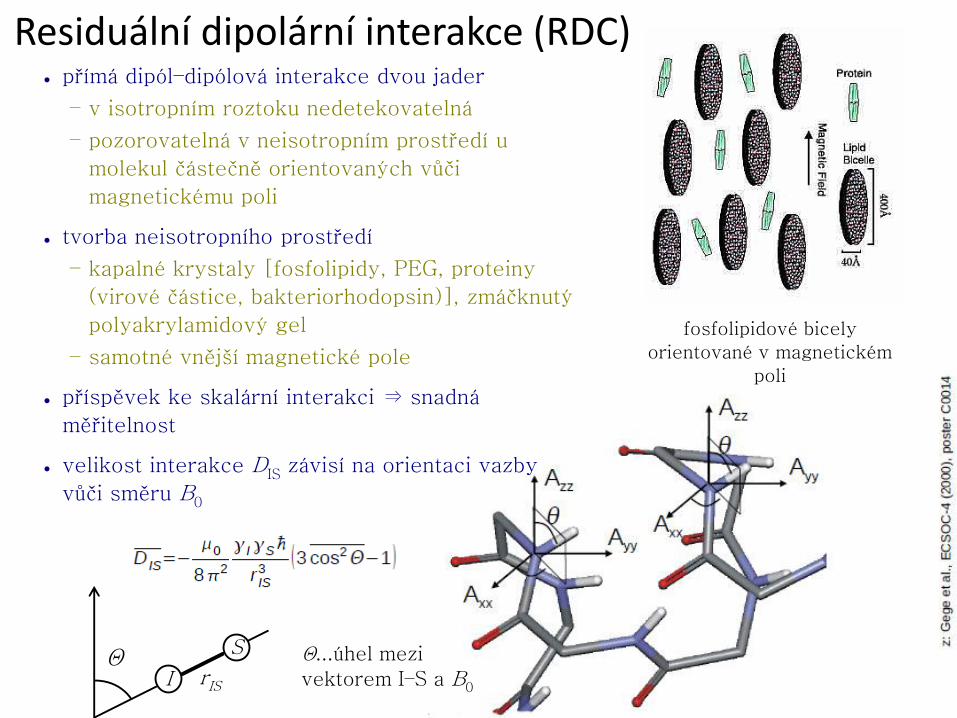
Využívaná jádra v biomakromolekulách
+ vysoké přirozené zastoupení
+ vysoká citlivost (1,00)
− malá disperse chemických posunů (cca. 15 ppm)
+ velká disperse chemických posunů (cca. 210 ppm)
− nízké přirozené zastoupení (1,11 %)
− nízká citlivost (1,76.10−4); po 100% isotopovém obohacení 1,59.10−2
• střední disperse chemických posunů (cca. 30 ppm)
− nízké přirozené zastoupení (0,37 %)
− nízká citlivost (3,85.10−6); po 100% isotopovém obohacení 1,04.10−3
• speciální účely
1H
13C
15N
2H
Zdroje Proteinů
• Původní organismus • + přirozená forma včetně všech modifikací • - malé množství, drahé, nemožnost izotopového značení, etické problémy,
složitá izolace,…. • Syntéza proteinu v mikroorganismech (E. coli, P. pastoris) • + levné, velký výtěžek proteinu, snadné uniformní izotopové značení • - možné problémy s modifikacemi postranních řetězců • Chemická syntéza • + velké možnosti izotopového značení, rychlé, vhodné pro toxické proteiny • - drahé, menší výtěžky, omezení maximální velikosti, problémy se
správným sbalením proteinu • In-vitro translace • + vhodné pro toxické proteiny, možnost selektivního izotopového značení • - drahé, posttranslační modifikace
Specifika řešení struktury proteinů pomocí NMR ● měření ve fyziologickém prostředí, možnost úpravy fyzikálně-chemických vlastností
prostředí
● sledování průběhu biochemických dějů
● vysoce selektivní odezva na úrovni atomů
Čím jsme omezeni:
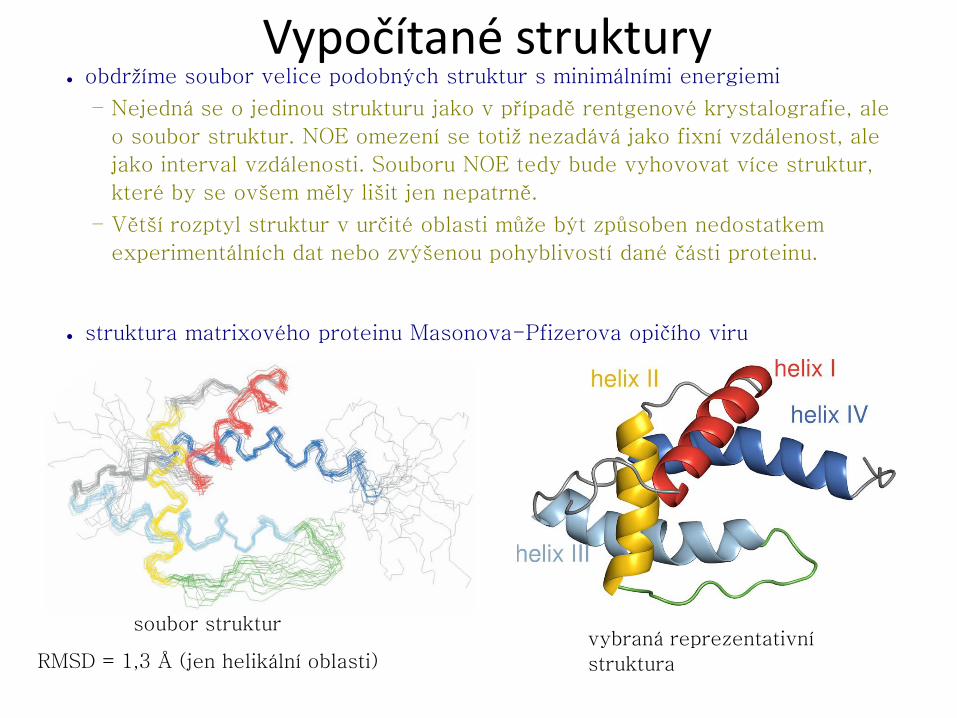
● velikost molekuly (ovlivnění T2)
– do 10 kDa (≡10 kg mol−1) [< 70 AA]
□ lze řešit přímo kombinací COSY, TOCSY a NOESY experimentů
– do 20 kDa [< 180 AA]
□ nutné 100% isotopové obohacení 13C a 15N
– do ~100 kDa
□ 100% isotopové obohacení 13C, 15N a částečné nebo úplné obohacení 2H (odstranění 1H jako hlavního zdroje rychlé relaxace 13C)
– větší proteiny
□ přístupný pouze hrubý „náhled“ na celkovou strukturu, sekundární struktura
● koncentrace vzorku
– alespoň 0,2 mM
● dlouhodobá stabilita vzorku
– několik týdnů
Vzorek proteinu pro NMR měření ● nutno dosáhnout koncentrace proteinu alespoň 0,2–
0,5 mmol l−1 (obecně více = lépe)
– používá se filtrace přes membrány s mikropóry
(protein zadržen) nebo lyofilizace a opětovné
rozpuštění
● úprava pH pufrem (pH typicky 4–8)
– vyšší pH by způsobilo rychlou výměnu
amidických vodíků s molekulami vody a ztrátu
signálů
● přidání redukčních činidel (R–SH)
– zabránění oxidace cysteinů a následného
vysrážení vzorku
● přidání 5–10 % D2O
– referenční jádro pro spektrometr (lock) roztok vzorku
250–300 µl
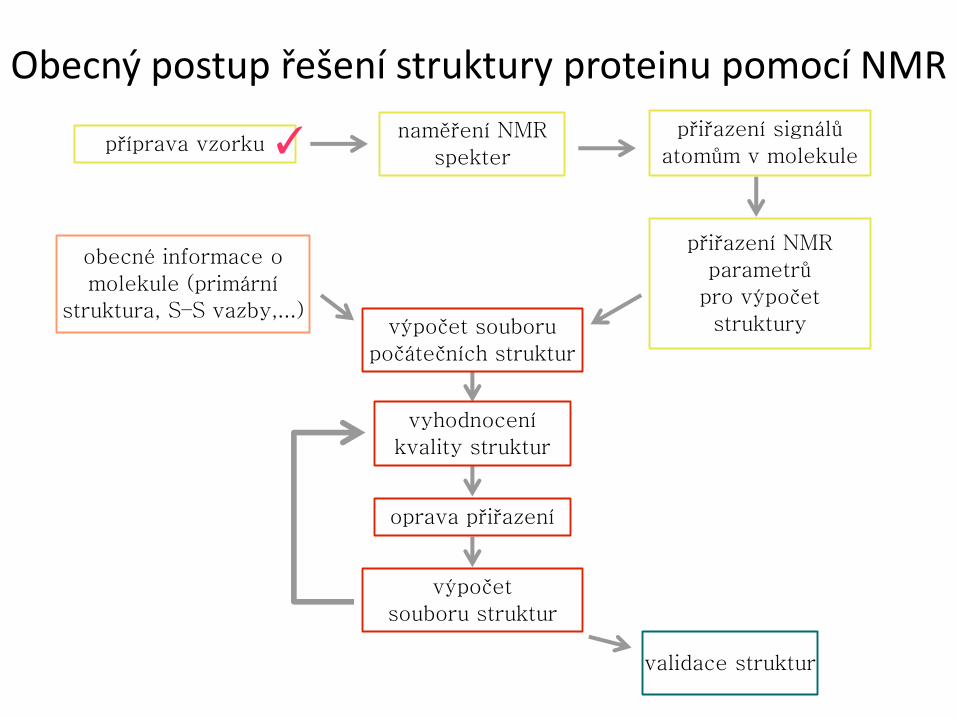
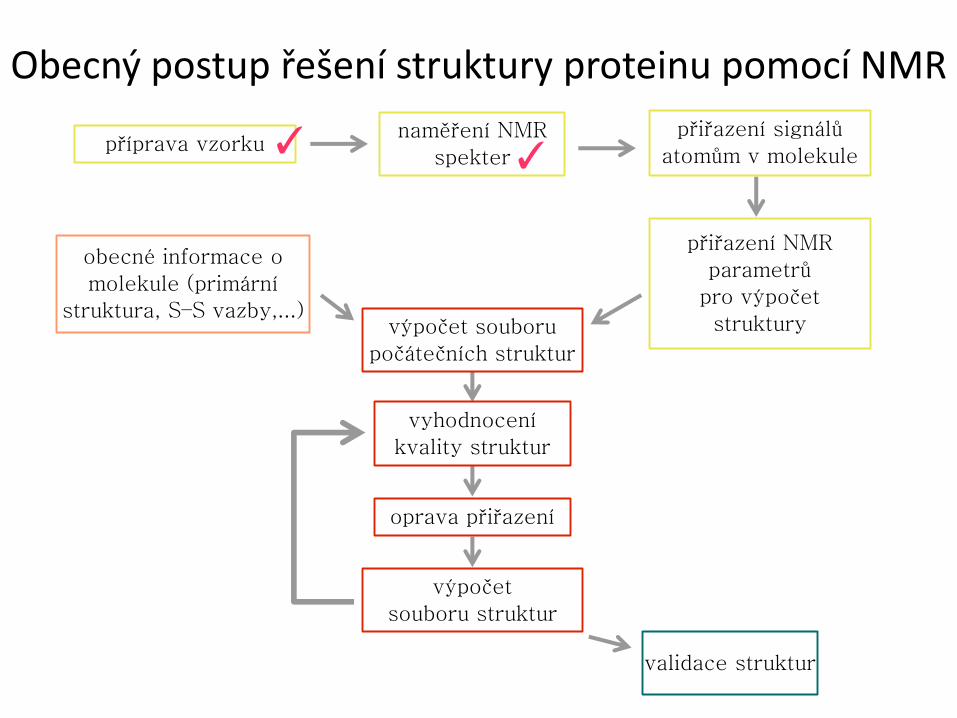
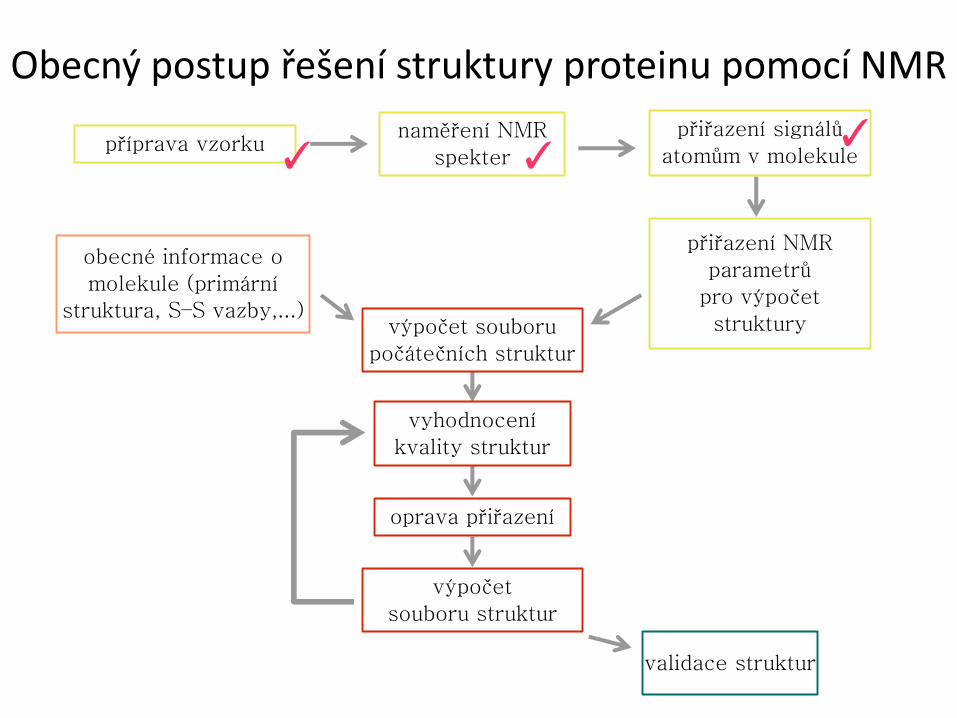
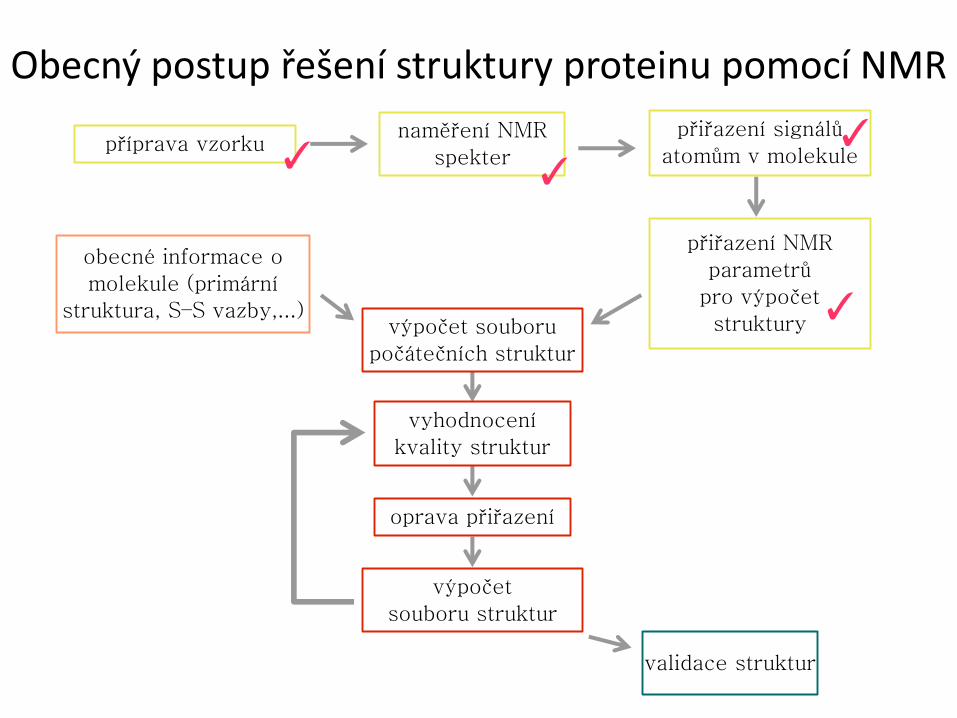
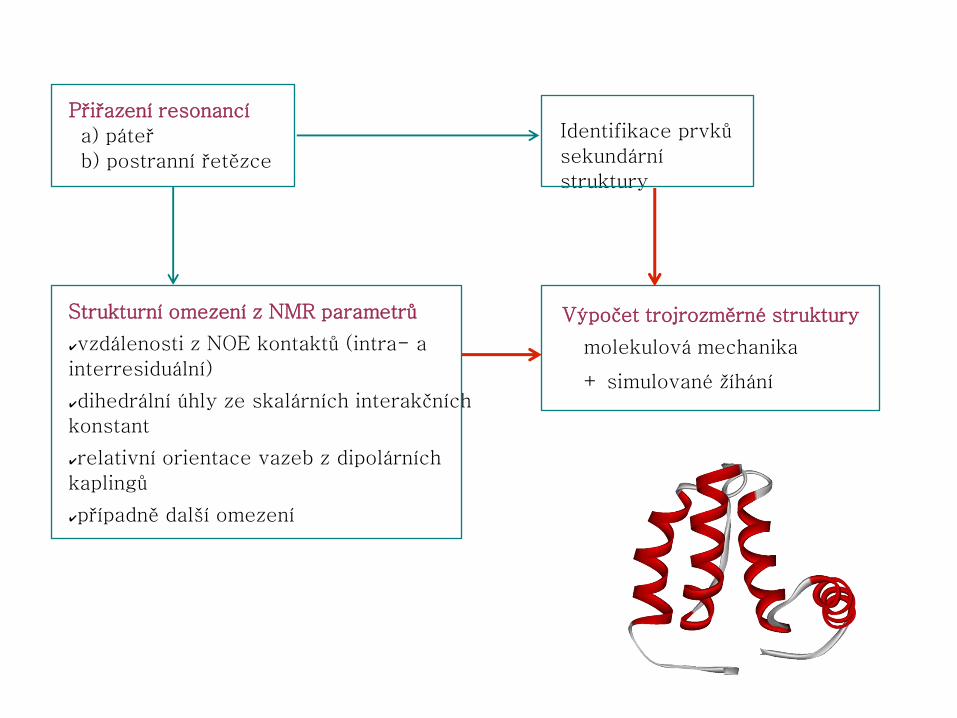
Obecný postup řešení struktury proteinu pomocí NMR
příprava vzorku naměření NMR
spekter
přiřazení signálů
atomům v molekule
přiřazení NMR
parametrů
pro výpočet
struktury
obecné informace o
molekule (primární
struktura, S–S vazby,...) výpočet souboru
počátečních struktur
vyhodnocení
kvality struktur
oprava přiřazení
výpočet
souboru struktur
validace struktur
✓
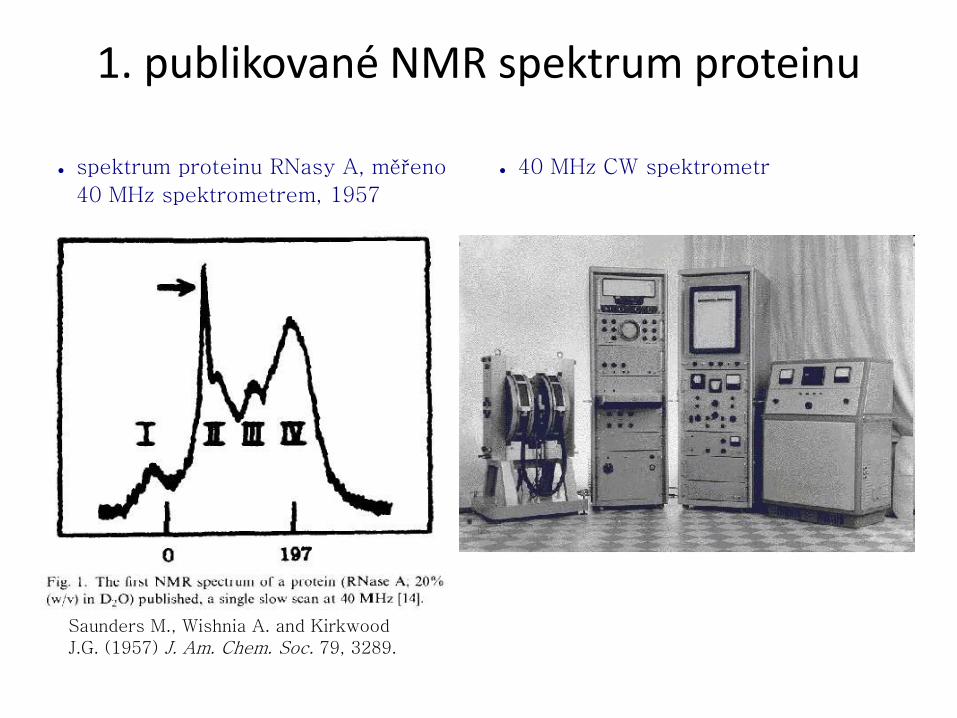
1. publikované NMR spektrum proteinu
● spektrum proteinu RNasy A, měřeno
40 MHz spektrometrem, 1957
● 40 MHz CW spektrometr
Saunders M., Wishnia A. and Kirkwood
J.G. (1957) J. Am. Chem. Soc. 79, 3289.
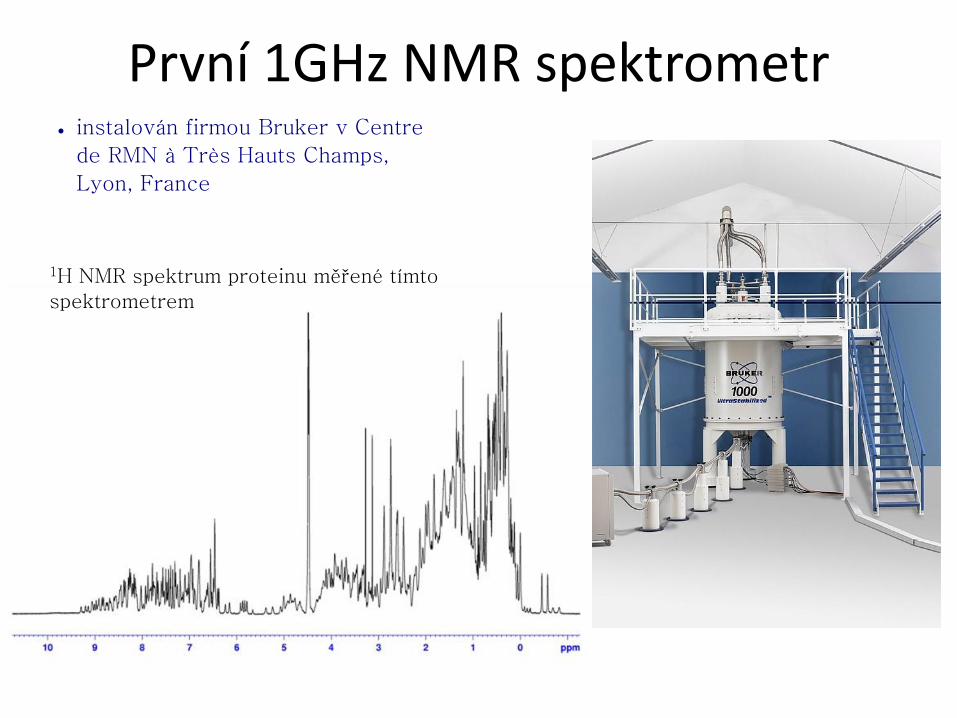
První 1GHz NMR spektrometr ● instalován firmou Bruker v Centre
de RMN à Très Hauts Champs,
Lyon, France
1H NMR spektrum proteinu měřené tímto
spektrometrem
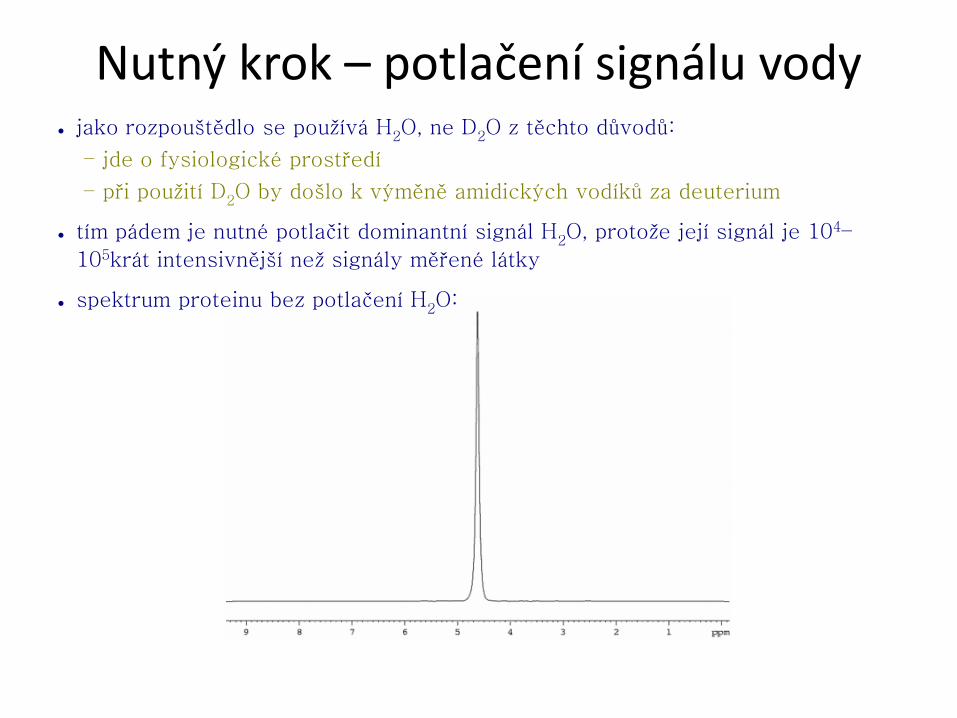
Nutný krok – potlačení signálu vody ● jako rozpouštědlo se používá H2O, ne D2O z těchto důvodů:
– jde o fysiologické prostředí
– při použití D2O by došlo k výměně amidických vodíků za deuterium
● tím pádem je nutné potlačit dominantní signál H2O, protože její signál je 104–
105krát intensivnější než signály měřené látky
● spektrum proteinu bez potlačení H2O:
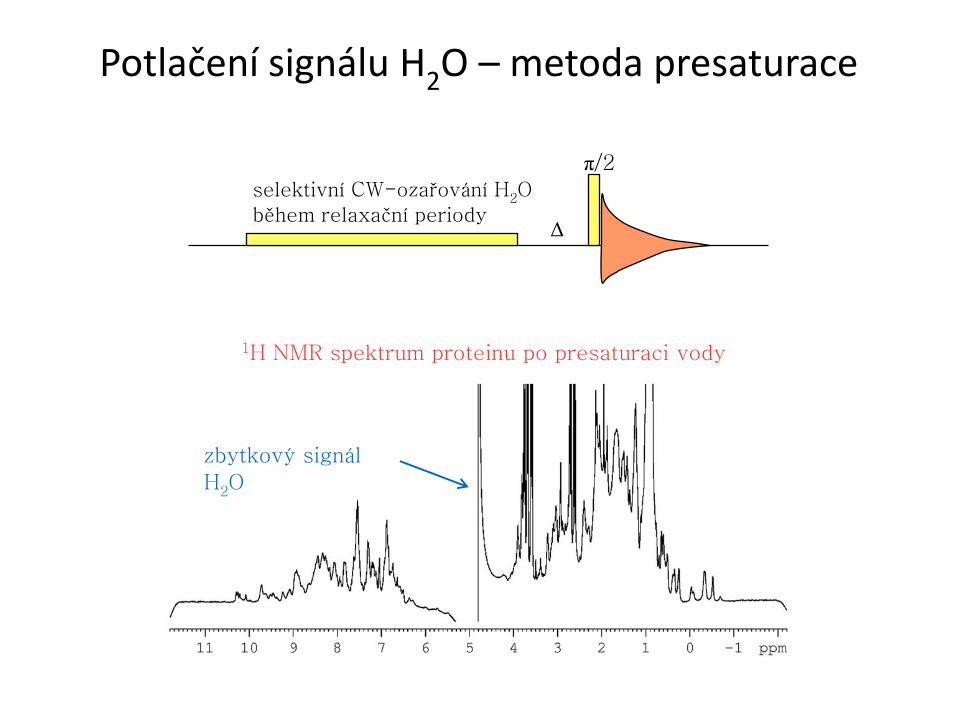
Potlačení signálu H2O – metoda presaturace
selektivní CW-ozařování H2O
během relaxační periody
π/2
Δ
zbytkový signál
H2O
1H NMR spektrum proteinu po presaturaci vody
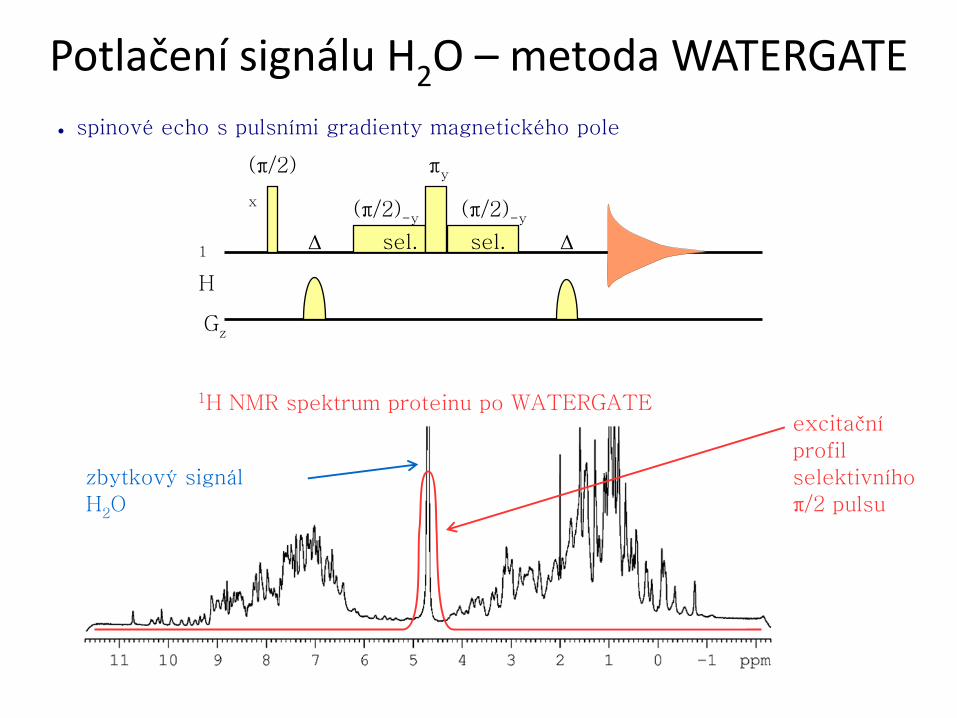
● spinové echo s pulsními gradienty magnetického pole
Potlačení signálu H2O – metoda WATERGATE
(π/2)-y
1
H
Gz
Δ
πy (π/2)
x
sel. sel. Δ
(π/2)-y
zbytkový signál
H2O
1H NMR spektrum proteinu po WATERGATE excitační
profil
selektivního
π/2 pulsu
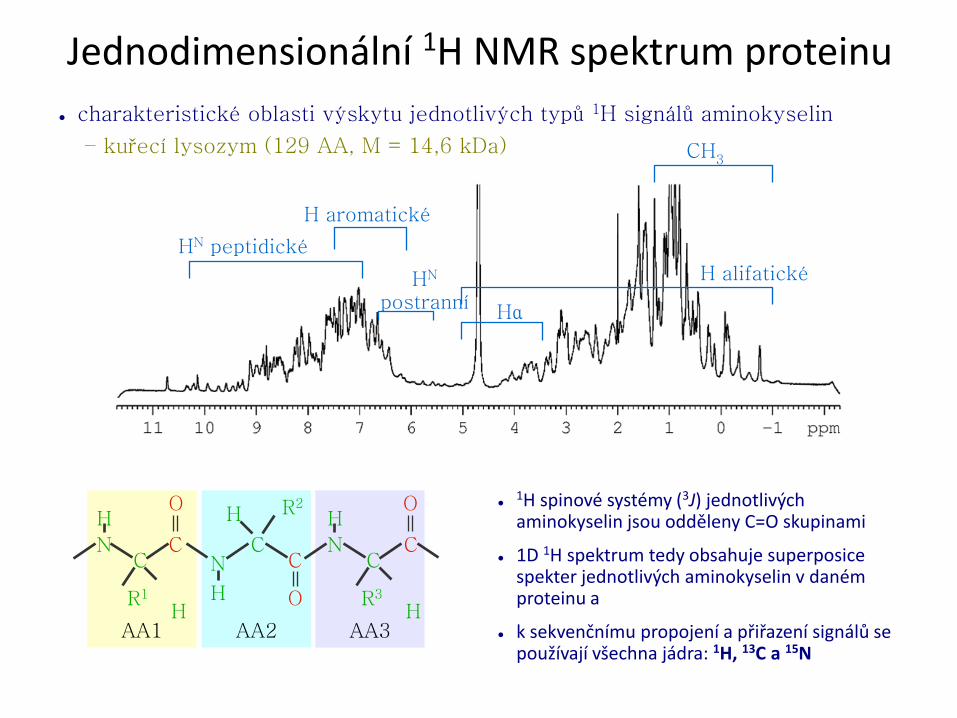
● charakteristické oblasti výskytu jednotlivých typů 1H signálů aminokyselin
– kuřecí lysozym (129 AA, M = 14,6 kDa)
H alifatické
HN peptidické
H aromatické
HN
postranní Hα
CH3
Jednodimensionální 1H NMR spektrum proteinu
AA3 AA2 AA1
C C N C
N C
C
O H
H H R2
O
H R1
H R3
N
H
C
O
●1H spinové systémy (3J) jednotlivých aminokyselin jsou odděleny C=O skupinami
● 1D 1H spektrum tedy obsahuje superposice spekter jednotlivých aminokyselin v daném proteinu a
● k sekvenčnímu propojení a přiřazení signálů se používají všechna jádra: 1H, 13C a 15N
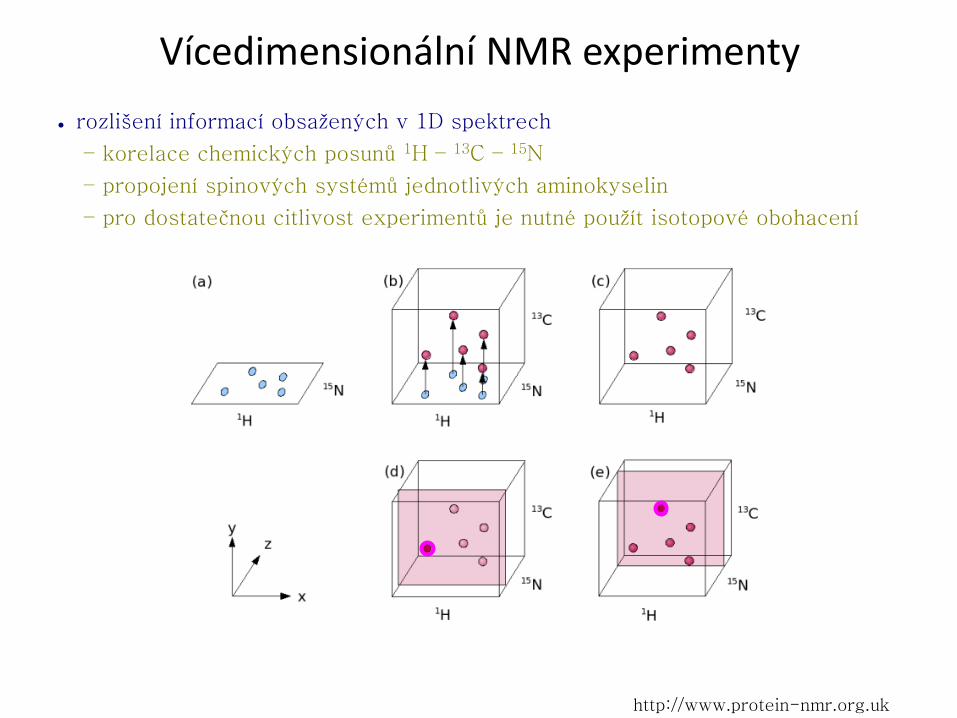
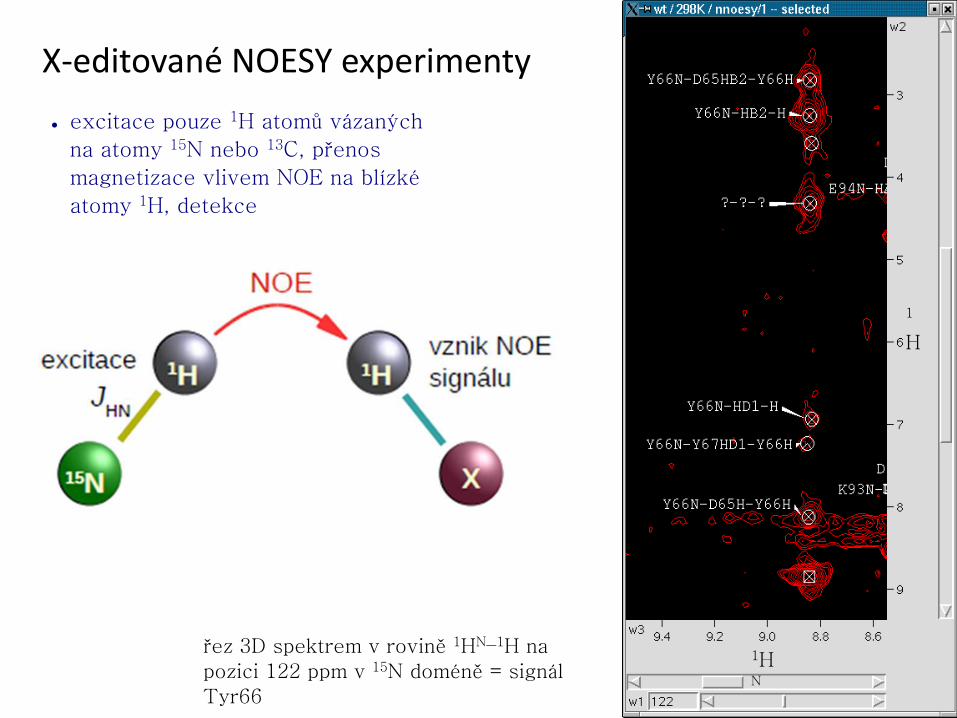
Vícedimensionální NMR experimenty
● rozlišení informací obsažených v 1D spektrech
– korelace chemických posunů 1H – 13C – 15N
– propojení spinových systémů jednotlivých aminokyselin
– pro dostatečnou citlivost experimentů je nutné použít isotopové obohacení
http://www.protein-nmr.org.uk
1H
(ppm)
1H
(ppm)
15N
(ppm
)
15N
(ppm
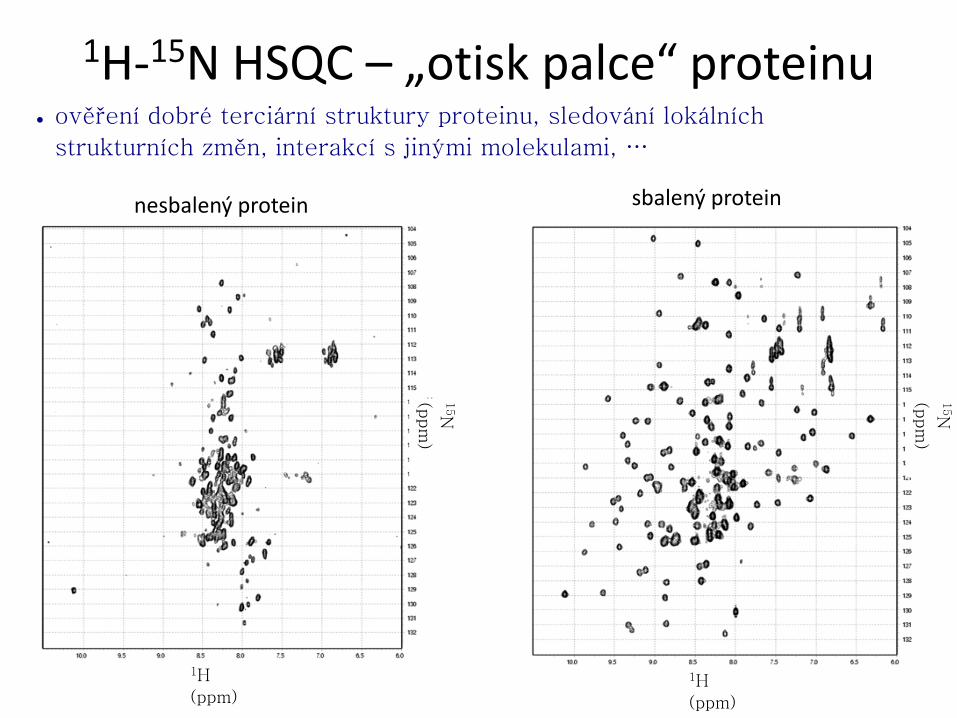
) 1H-15N HSQC – „otisk palce“ proteinu
● ověření dobré terciární struktury proteinu, sledování lokálních
strukturních změn, interakcí s jinými molekulami, …
nesbalený protein sbalený protein
Obecný postup řešení struktury proteinu pomocí NMR
příprava vzorku naměření NMR
spekter
přiřazení signálů
atomům v molekule
přiřazení NMR
parametrů
pro výpočet
struktury
obecné informace o
molekule (primární
struktura, S–S vazby,...) výpočet souboru
počátečních struktur
vyhodnocení
kvality struktur
oprava přiřazení
výpočet
souboru struktur
validace struktur
✓ ✓
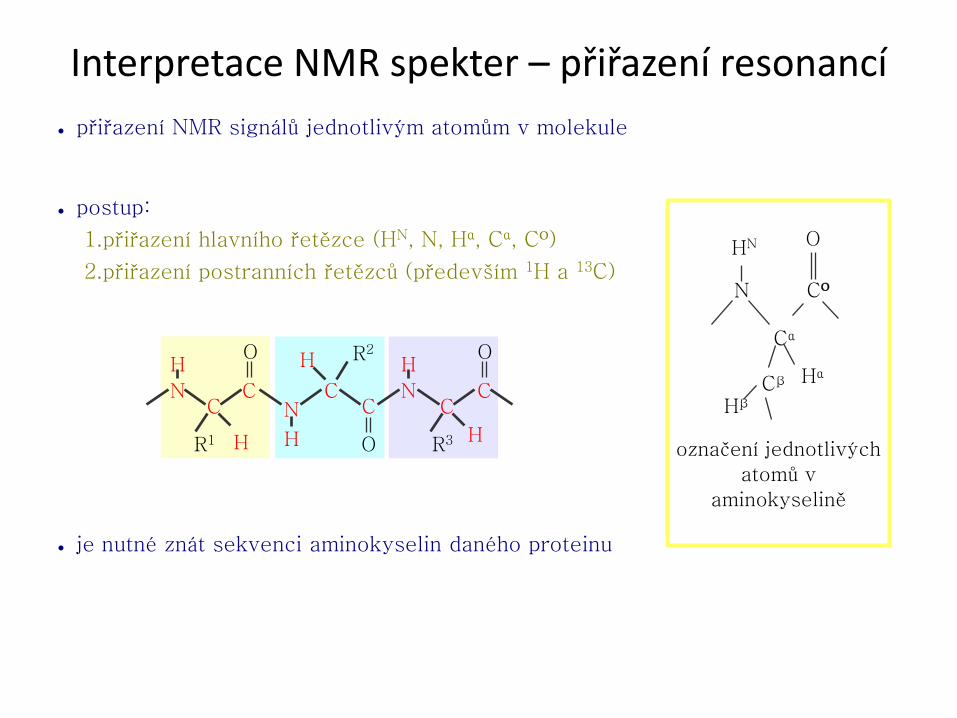
Interpretace NMR spekter – přiřazení resonancí
● přiřazení NMR signálů jednotlivým atomům v molekule
● postup:
1.přiřazení hlavního řetězce (HN, N, Hα, Cα, CO)
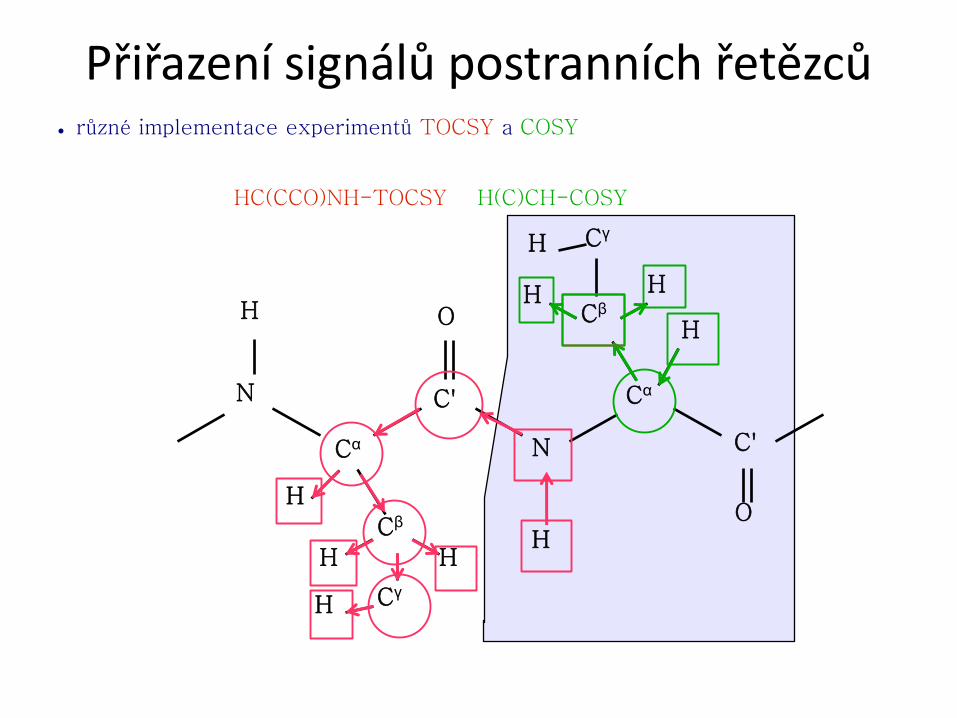
2.přiřazení postranních řetězců (především 1H a 13C)
● je nutné znát sekvenci aminokyselin daného proteinu
C C N C
N C
C
O H
H H R2
O
H
R1
H
R3
N
H
C O
CO
Cα
O
Hα Cβ
N
HN
Hβ
označení jednotlivých
atomů v
aminokyselině
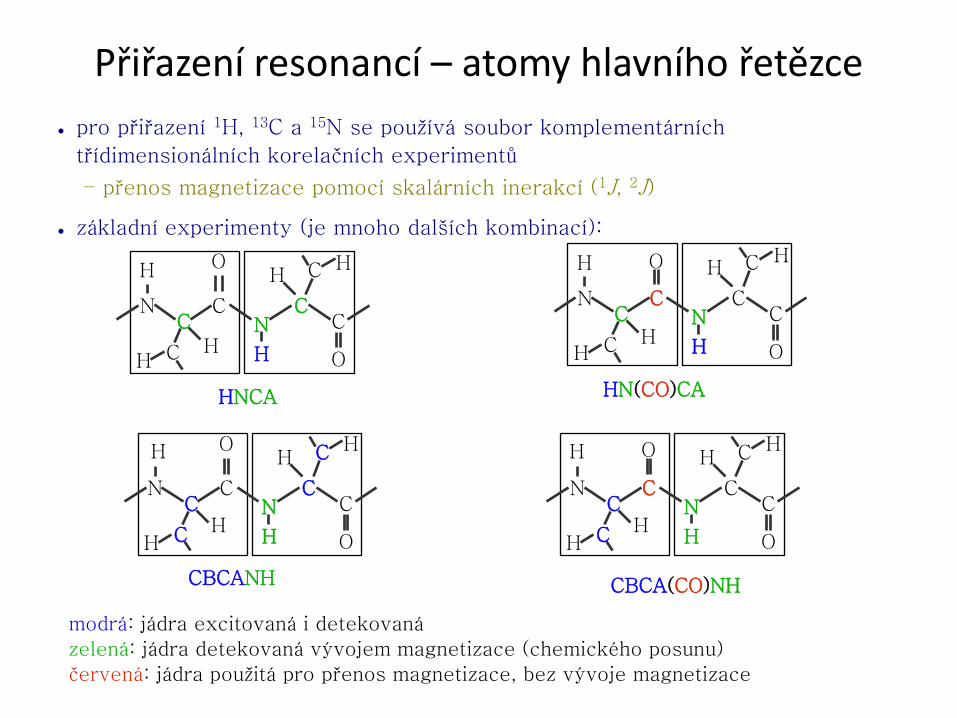
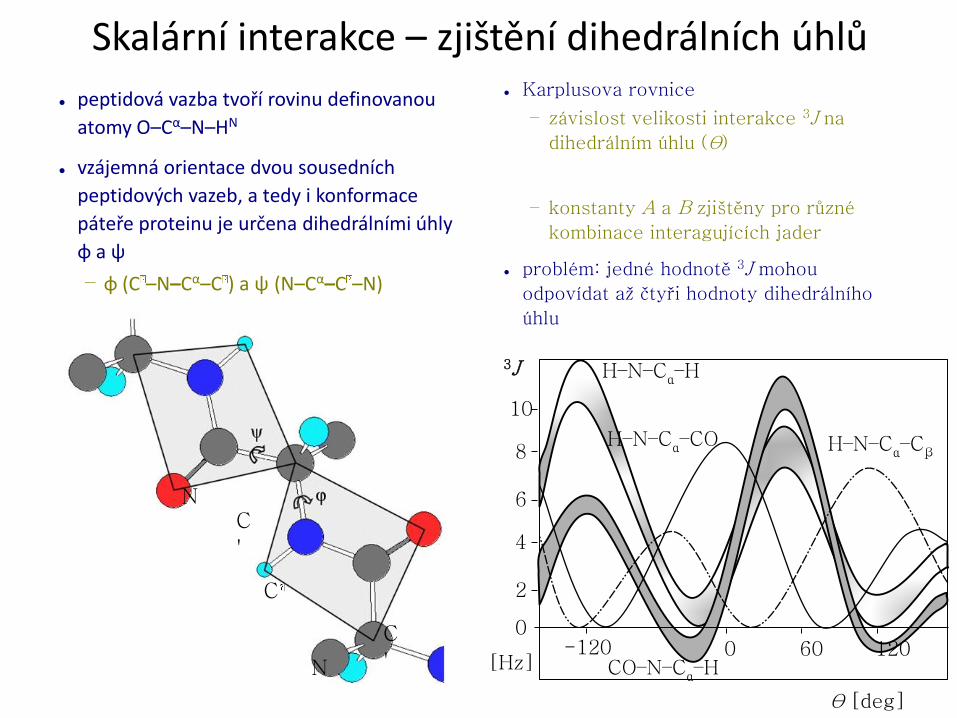
Přiřazení resonancí – atomy hlavního řetězce
● pro přiřazení 1H, 13C a 15N se používá soubor komplementárních
třídimensionálních korelačních experimentů
– přenos magnetizace pomocí skalárních inerakcí (1J, 2J)
● základní experimenty (je mnoho dalších kombinací):
C C
N C
C
O H
H C
O
H C
N
H
H
H
HNCA
C C
N C
C
O H
H C
O
H
C
N
H
H
H
HN(CO)CA
C C
N C
C
O H
H C O
H
C
N
H
H
H
CBCANH
C C
N C
C
O H
H C
O
H
C
N
H
H
H
CBCA(CO)NH
modrá: jádra excitovaná i detekovaná
zelená: jádra detekovaná vývojem magnetizace (chemického posunu)
červená: jádra použitá pro přenos magnetizace, bez vývoje magnetizace
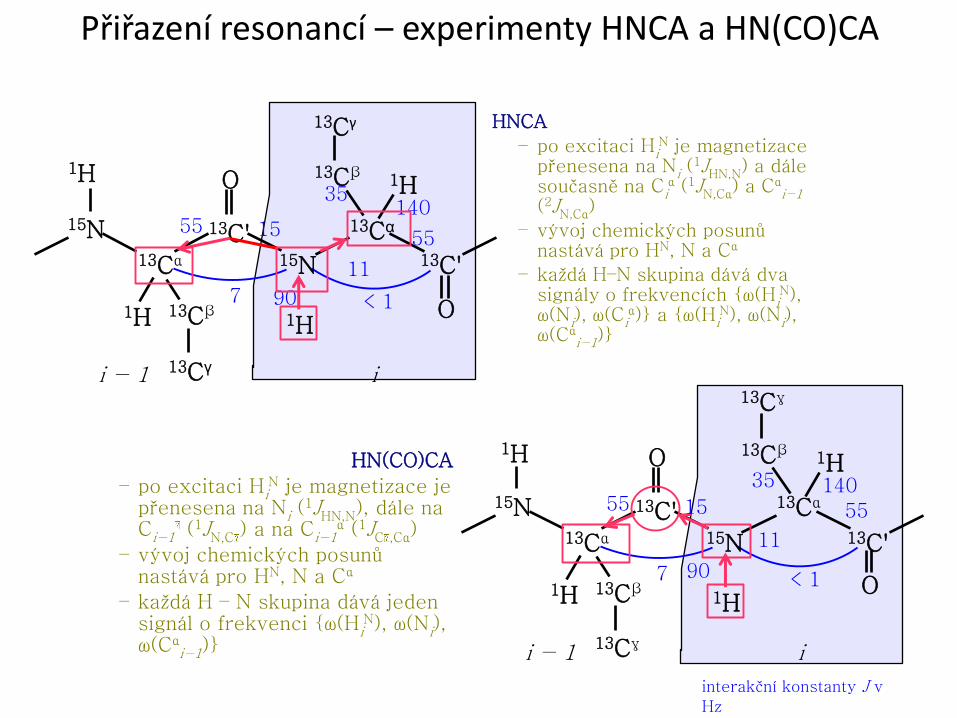
Přiřazení resonancí – experimenty HNCA a HN(CO)CA
13Cα
13C' 15N
13Cα
13C'
15N
O
O 1H
1H 13Cβ
13Cγ
1H
1H 13Cβ
13Cγ
90
11
15
7
55 140
55
35
i − 1 i
< 1
13Cα
13C' 15N
13Cα
13C'
15N
O
O 1H
1H 13Cβ
13Cγ
1H
1H 13Cβ
13Cγ
90
11
15
7
55 140
55
35
i − 1 i
< 1
interakční konstanty J v
Hz
HN(CO)CA
– po excitaci HiN je magnetizace je
přenesena na Ni (1JHN,N), dále na
Ci−1 (1JN,C ) a na Ci−1α (1JC ,Cα)
– vývoj chemických posunů nastává pro HN, N a Cα
– každá H – N skupina dává jeden signál o frekvenci {ω(Hi
N), ω(Ni), ω(Cα
i−1)}
HNCA
– po excitaci HiN je magnetizace
přenesena na Ni (1JHN,N) a dále
současně na Ciα (1JN,Cα) a Cα
i−1 (2JN,Cα)
– vývoj chemických posunů nastává pro HN, N a Cα
– každá H–N skupina dává dva signály o frekvencích {ω(Hi
N), ω(Ni), ω(Ci
α)} a {ω(HiN), ω(Ni),
ω(Cαi−1)}
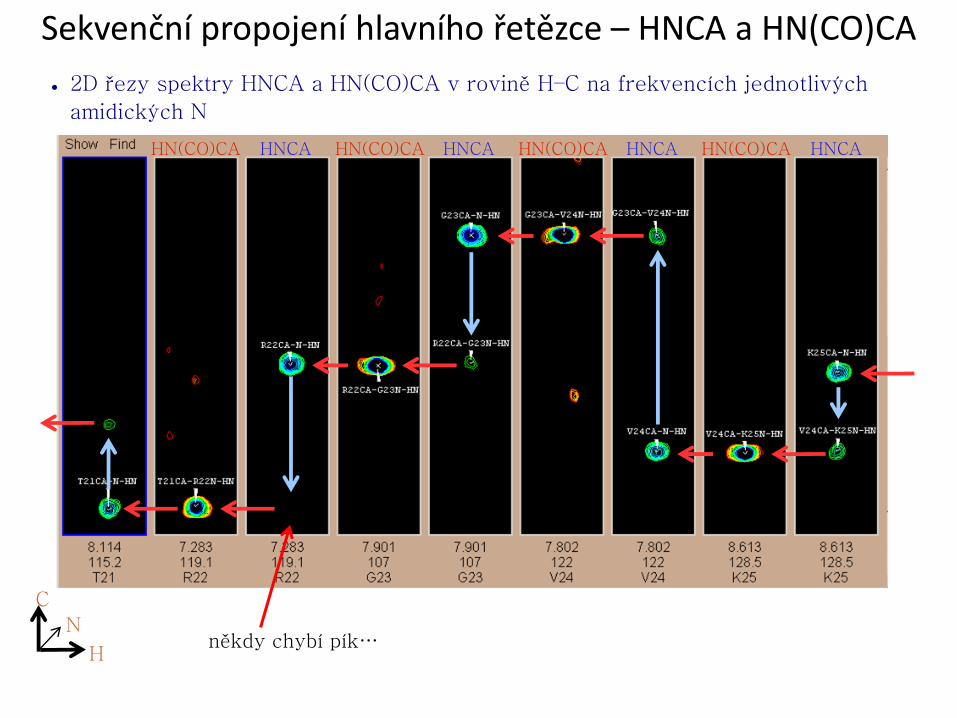
Sekvenční propojení hlavního řetězce – HNCA a HN(CO)CA
● 2D řezy spektry HNCA a HN(CO)CA v rovině H–C na frekvencích jednotlivých